CPPTRAJ for Analysis
Learning Objectives
Activities
Trajectory Analysis with CPPTRAJ
Input files on the cluster are located in the following directory:
working_directory/input/CPPTRAJ
Input files are also available here:
CPPTRAJ Inputs.
Reference output files are also available here:
CPPTRAJ Outputs.
Note
The following commands are intended for users running on the HPC environment. If you are using a local machine, you may already have CPPTRAJ installed and do not need to load modules or activate a software environment.
To load the required software on the HPC system, enter the following commands in your terminal:
module load Amber/22.0-foss-2021b-AmberTools-22.3-CUDA-11.4.1
module load Grace
The Grace module provides xmgrace, which will be used later in this tutorial to visualize analysis results.
CPPTRAJ Roe and Cheatham III[1] is a valuable asset to complement your computational toolkit, as this AMBER program can be used to achieve a more rigorous analysis of MD trajectories. In this section, you will learn how to use CPPTRAJ to wrap trajectories, calculate RMSDs/RMSFs, and find an average structure from a trajectory. For this activity, you will be given multiple CPPTRAJ input files. Each input file contains lines of commands for CPPTRAJ, but they can each be individually submitted in the interactive command line of CPPTRAJ. The interactive command line of CPPTRAJ can be accessed by typing into your terminal:
cpptraj
Which will open up the interactive command line seen below:
CPPTRAJ: Trajectory Analysis. V4.26.4 (AmberTools V20.00)
___ ___ ___ ___
| \/ | \/ | \/ |
_|_/\_|_/\_|_/\_|_
| Date/time: 00/00/26 00:00:00
| Available memory: 54.268 GB
To exit CPPTRAJ, simply type: quit.
Wrapping Trajectories
We will once again return to the MTR1 system.
cd working_dir/input/CPPTRAJ
Before you start the analysis of a trajectory, you must first wrap the system. Wrapping a trajectory means adjusting the positions of the molecules in a simulation so that they can stay within your defined simulation box. If you were to open an unwrapped trajectory in VMD, you would see that the molecules within the system can appear to look separated, drifting away or blown out due to the periodic boundary conditions. Therefore, it can be hard to gain an accurate interpretation of what is true behavior displayed in your system. If you are wanting to visually inspect your system, wrapping should be the first step prior to trajectory analysis to avoid confusion.
Wrapping is one of CPPTRAJ’s most utilized roles. CPPTRAJ can wrap both restart files and trajectory files. In this case, you will be wrapping a MTR1 trajectory file. First, you must decide on a center in which to wrap everything around. It is a good practice to center your system around the solute, or you can be more specific and choose the catalytic center residues or a ligand.
Access the CPPTRAJ interactive command line:
cpptraj
Like VMD, you must load the topology file first, followed by the trajectory or restart file. CPPTRAJ’s command to load a topology file is parm.
> parm MTR1_prot-run.parm7
Note
You could also try typing the following into your terminal to load the topology file:
cpptraj -p MTR1_prot-run.parm7
Once CPPTRAJ loads, you’ll be at the interactive prompt. Load your trajectory file using the trajin command:
trajin MTR1_prot-run.nc
Note
Windows Users: you can load the parm and trajectory file and use the trajout command to create a DCD trajectory file.
trajout MTR1_prot-run.dcd
Now apply the wrapping commands. The basic wrapping workflow uses the reference, center and image commands. The reference command specifies the reference structure for centering and imaging. The center command moves the center of mass of the specified group to the origin, and the image command re-images all other molecules into the primary periodic box based on this centered group.
For MTR1, you will be centering around the solute. A typical mask for the solute can be described as :
solute='!@H= & !:MG,NA,CL,PB,ZN,MN,WAT'
This mask selects all atoms that are not hydrogens, water, nor magnesium, sodium, chlorine, or lead ions. Use this mask to center your system around the solute. Essentially, it tells cpptraj to center the system around the heavy atoms of the solute. This is a more generalized mask. You can create a more specific mask to center around the catalytic residues or a ligand. For example:
solute='!@H= & !:MG,NA,CL,PB,ZN,MN,WAT & :1-69'
Note
The symbols used in the mask have specific meanings:
@— Refers toatom names:— Refers toresidue names!— LogicalNOT(exclude)&— LogicalAND=— Wildcard (matches anything that begins with the preceding text)
Type the following commands into the CPPTRAJ interactive command line one by one to wrap your trajectory:
reference MTR1_prot-run.rst7
center '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT' mass origin
autoimage '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT'
center '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT' mass reference
Note
The atom selection syntax in CPPTRAJ might take some time getting used to.
massindicates that the centering should be done based on the center of mass of the atom selection.autoimageautomatically re-image coordinates.originmeans that the center of mass will be moved to the origin (0,0,0) of the coordinate system.
To write out the wrapped trajectory:
trajout centered.MTR1_prot-run.nc
trajout centered.MTR1_prot-run.dcd dcd
Note
Windows Users: use the trajout command to create a DCD trajectory file.
Execute the commands by typing:
run
or
go
The wrapped trajectory will now be saved and in your working directory. Once the job is finished running, type quit to exit CPPTRAJ and the interactive command line. You can visualize this in VMD to see that the system appears intact with all molecules properly positioned within the box around your solute of interest, or in this case, MTR1.
Your cpptraj interactive command line should look something like this:
CPPTRAJ: Trajectory Analysis. V4.26.4 (AmberTools V20.00)
___ ___ ___ ___
| \/ | \/ | \/ |
_|_/\_|_/\_|_/\_|_
| Date/time: 07/07/26 15:58:49
| Available memory: 3.037 GB
> parm MTR1_prot-run.parm7
[parm MTR1_prot-run.parm7]
Reading 'MTR1_prot-run.parm7' as Amber Topology
Radius Set: modified Bondi radii (mbondi)
> trajin MTR1_prot-run.nc
[trajin MTR1_prot-run.nc]
Reading 'MTR1_prot-run.nc' as Amber NetCDF
> reference MTR1_prot-run.rst7
[reference MTR1_prot-run.rst7]
Reading 'MTR1_prot-run.rst7' as Amber Restart
Setting active reference for distance-based masks: 'MTR1_prot-run.rst7'
> center '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT' mass origin
[center '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT' mass origin]
CENTER: Centering coordinates using center of mass of atoms in mask (!@H* & !:MG,NA,CL,PB,ZN,MN,WAT) to
coordinate origin.
> autoimage '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT'
[autoimage '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT']
AUTOIMAGE: To box center based on center of mass, anchor mask is [!@H= & !:MG,NA,CL,PB,ZN,MN,WAT]
> center '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT' mass reference
[center '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT' mass reference]
CENTER: Centering coordinates using center of mass of atoms in mask (!@H* & !:MG,NA,CL,PB,ZN,MN,WAT) to
center of mask (!@H* & !:MG,NA,CL,PB,ZN,MN,WAT) in reference 'MTR1_prot-run.rst7'.
> trajout centered.MTR1_prot-run.nc
[trajout centered.MTR1_prot-run.nc]
Writing 'centered.MTR1_prot-run.nc' as Amber NetCDF
> trajout centered.MTR1_prot-run.dcd dcd
[trajout centered.MTR1_prot-run.dcd dcd]
Writing 'centered.MTR1_prot-run.dcd' as Charmm DCD
> go
[go]
---------- RUN BEGIN -------------------------------------------------
PARAMETER FILES (1 total):
0: MTR1_prot-run.parm7, 72443 atoms, 17745 res, box: Trunc. Oct., 17678 mol, 17520 solvent
INPUT TRAJECTORIES (1 total):
0: 'MTR1_prot-run.nc' is a NetCDF AMBER trajectory with coordinates, time, box, Parm MTR1_prot-run.parm7 (Trunc. Oct. box) (reading 100 of 100)
Coordinate processing will occur on 100 frames.
REFERENCE FRAMES (1 total):
0: MTR1_prot-run.rst7:1
Active reference frame for distance-based masks is 'Cpptraj Generated Restart'
OUTPUT TRAJECTORIES (2 total):
'centered.MTR1_prot-run.nc' (100 frames) is a NetCDF AMBER trajectory
'centered.MTR1_prot-run.dcd' (100 frames) is a CHARMM DCD file (coords) Little Endian 32 bit
BEGIN TRAJECTORY PROCESSING:
.....................................................
ACTION SETUP FOR PARM 'MTR1_prot-run.parm7' (3 actions):
0: [center '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT' mass origin]
Mask [!@H* & !:MG,NA,CL,PB,ZN,MN,WAT] corresponds to 1459 atoms.
1: [autoimage '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT']
Original box is truncated octahedron, turning on 'familiar'.
Anchoring on atoms selected by mask '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT'
Mask [!@H* & !:MG,NA,CL,PB,ZN,MN,WAT] corresponds to 1459 atoms.
2 molecules are fixed to anchor: 1 2
17676 molecules are mobile.
2: [center '!@H= & !:MG,NA,CL,PB,ZN,MN,WAT' mass reference]
Mask [!@H* & !:MG,NA,CL,PB,ZN,MN,WAT] corresponds to 1459 atoms.
.....................................................
ACTIVE OUTPUT TRAJECTORIES (2):
centered.MTR1_prot-run.nc (coordinates, time, box)
centered.MTR1_prot-run.dcd (coordinates, time, box)
----- MTR1_prot-run.nc (1-100, 1) -----
0% 10% 20% 30% 40% 51% 61% 71% 81% 91% 100% Complete.
Read 100 frames and processed 100 frames.
TIME: Avg. throughput= 86.0656 frames / second.
ACTION OUTPUT:
TIME: Analyses took 0.0000 seconds.
RUN TIMING:
TIME: Init : 0.0001 s ( 0.01%)
TIME: Trajectory Process : 1.1619 s ( 99.79%)
TIME: Action Post : 0.0000 s ( 0.00%)
TIME: Analysis : 0.0000 s ( 0.00%)
TIME: Data File Write : 0.0000 s ( 0.00%)
TIME: Other : 0.0023 s ( 0.00%)
TIME: Run Total 1.1643 s
---------- RUN END ---------------------------------------------------
> quit
[quit]
--------------------------------------------------------------------------------
To cite CPPTRAJ use:
Daniel R. Roe and Thomas E. Cheatham, III, "PTRAJ and CPPTRAJ: Software for
Processing and Analysis of Molecular Dynamics Trajectory Data". J. Chem.
Theory Comput., 2013, 9 (7), pp 3084-3095.
The commands that you just entered can be contained in a text file, called wrap.in, which can look like this:
parm MTR1_prot-run.parm7
trajin MTR1_prot-run.nc
reference MTR1_prot-run.rst7
center '!@H= & !:MG= & !:NA= & !:CL= & !:PB= & !:WAT=' mass origin
autoimage '!@H= & !:MG= & !:NA= & !:CL= & !:PB= & !:WAT='
center '!@H= & !:MG= & !:NA= & !:CL= & !:PB= & !:WAT=' mass reference
trajout centered.MTR1_prot-run.nc
trajout centered.MTR1_prot-run.dcd dcd
run
quit
Then, you would use the following to execture CPPTRAJ to run the commands in the text file:
cpptraj -i wrap.in
You can also create a more comprehensive bash script to automate the process of wrapping your trajectory. The script can look like this:
#!/bin/bash
# Set Variables #
parFile=MTR1_prot-run.parm7
restartFile=MTR1_prot-run.rst7
trajFile=MTR1_prot-run.nc
# Define Solute Mask #
solute='!@H= & !:MG= & !:NA= & !:CL= & !:PB= & !:WAT='
# Write CPPTRAJ input file #
cat <<EOF > wrap.in
parm ${parFile}
trajin ${trajFile}
reference ${restartFile}
center '${solute}' mass origin
autoimage '${solute}'
center '${solute}' mass reference
trajout centered.${trajFile}
run
quit
EOF
# Run input file to wrap trajectory #
cpptraj -i wrap.in > c.log
rm c.log
Run by:
chmod +x wrap.sh
./wrap.sh
Computing Root Mean Square Deviations (RMSD)
For the following, you will have to ssh into a remote server. To do this, type the following into your terminal:
ssh -X your_username@hyperion.sw.ehu.es
Now, you have already learned how to create RMSD plots using VMD. CPPTRAJ also has the capability to calculate RMSDs. We will use it to calculate RMSDs, RMSFs and average structures.
In directory you will find the input script called ctraj.RMSD_MTR1.in. It looks like this:
parm MTR1_prot-run.parm7
trajin centered.MTR1_prot-run.nc
rms ToFirst :1-69&!@H= first out RMSD_MTR1.dat mass
run
quit
Note
This is a standalone CPPTRAJ input file that can be run directly with cpptraj. It contains only CPPTRAJ commands.
This input file again follows a standard of inputting your topology file first, then your trajectory. The rms ToFirst :1-69&!@H= first out RMSD_MTR1.dat mass line tells cpptraj to do an RMSD calculation, saving the data set as ToFirst, using all non-hydrogen atoms in the residues 1 through 69. This residue selection is the entire nucleic molecule. The first is telling cpptraj to use the first frame in the trajectory as a reference, with out as the command to write the output to a file named RMSD_MTR1.dat. The mass command indicates a mass-weighted RMSD calculation. Submit this file as you did before:
cpptraj -i ctraj.RMSD_MTR1.in
Once the job is complete, the RMSD_MTR1.dat file will be in your directory.
Using xmgrace, open this file:
xmgrace RMSD_MTR1.dat
Which will pop up:
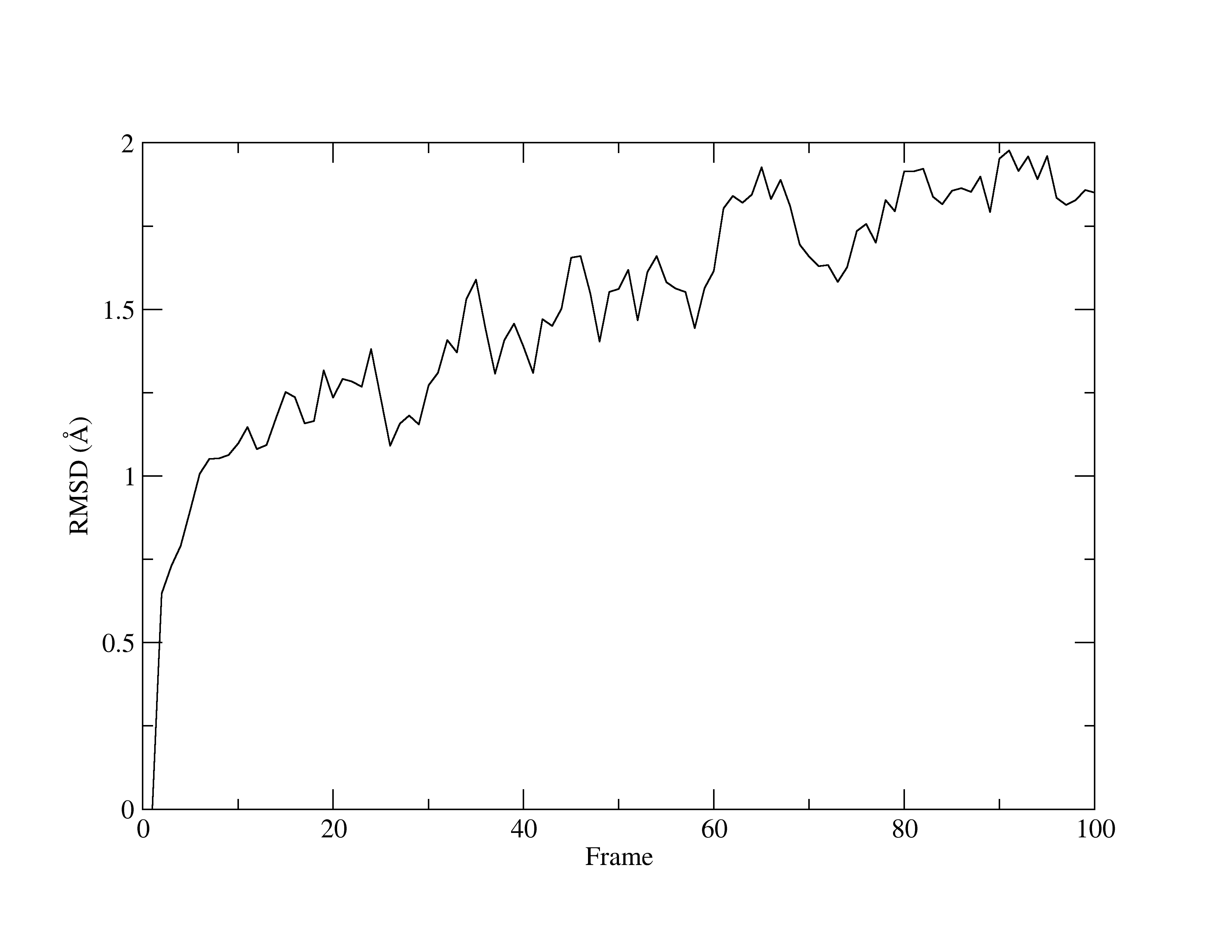
Figure 1. RMSD of MTR1.
With a quick glance of the graph, you will see that it is comparable to the same graph that was produced by VMD. Once again, the line fluctuates and has an upwards direction. Yet, given the very low values for RMSD, you can concur that the structure is stable.
Computing Root Mean Square Fluctuations (RMSF)
Next, you will learn to use CPPTRAJ to calculate a Root Mean Square Fluctuation (RMSF). Like RMSDs, RMSFs are important analytical measurements that provide insight into your system. RMSFs quantify the overall flexibility and mobility of each atom or residue within your system as the simulation progresses. RMSFs calculate how much the position of each atom in your selection deviates from its average position throughout the simulation time.
In your directory, find the input file, ctraj.RMSF_MTR1.in.
Open in VIM to see the script:
parm MTR1_prot-run.parm7
reference centered.MTR1_prot-run.rst7
trajin centered.MTR1_prot-run.nc
rms :1-69&!@H reference mass
atomicfluct out RMSF_MTR1.dat :1-69&!@H= byres
atomicfluct out RMSF_Bfact_MTR1.dat :1-69&!@H= byres bfactor
run
quit
The script is similar to the previous one, but there are two new lines containing the RMSF commands. atomicfluct out RMSF.dat :1-69&!@H= byres and atomicfluct out RMSF_Bfact.dat :1-69&!@H= byres bfactor. atomicfluct is the command for an RMSF calculation. As seen before, out directs CPPTRAJ to output the data into a file name of your choosing (here, it is RMSF.dat), followed by your atom selection. The keyword byres specifies the output to be per residue, which sums the fluctuations of all atoms in each residue. The other keyword seen, bfactor, which will calculate the B-factor of the MD trajectory. This is an additional analysis one can do to compare simulation to crystallographic data. A low B-factor is indicative of a well-ordered and stable structure, while a high b-factor suggests more movement and thereby, greater fluctuations.
Run the input file as you did before:
cpptraj -i ctraj.RMSF_MTR1.in
Open the RMSF_MTR1.dat by using xmgrace:
xmgrace RMSF_MTR1.dat
Which will look like this:
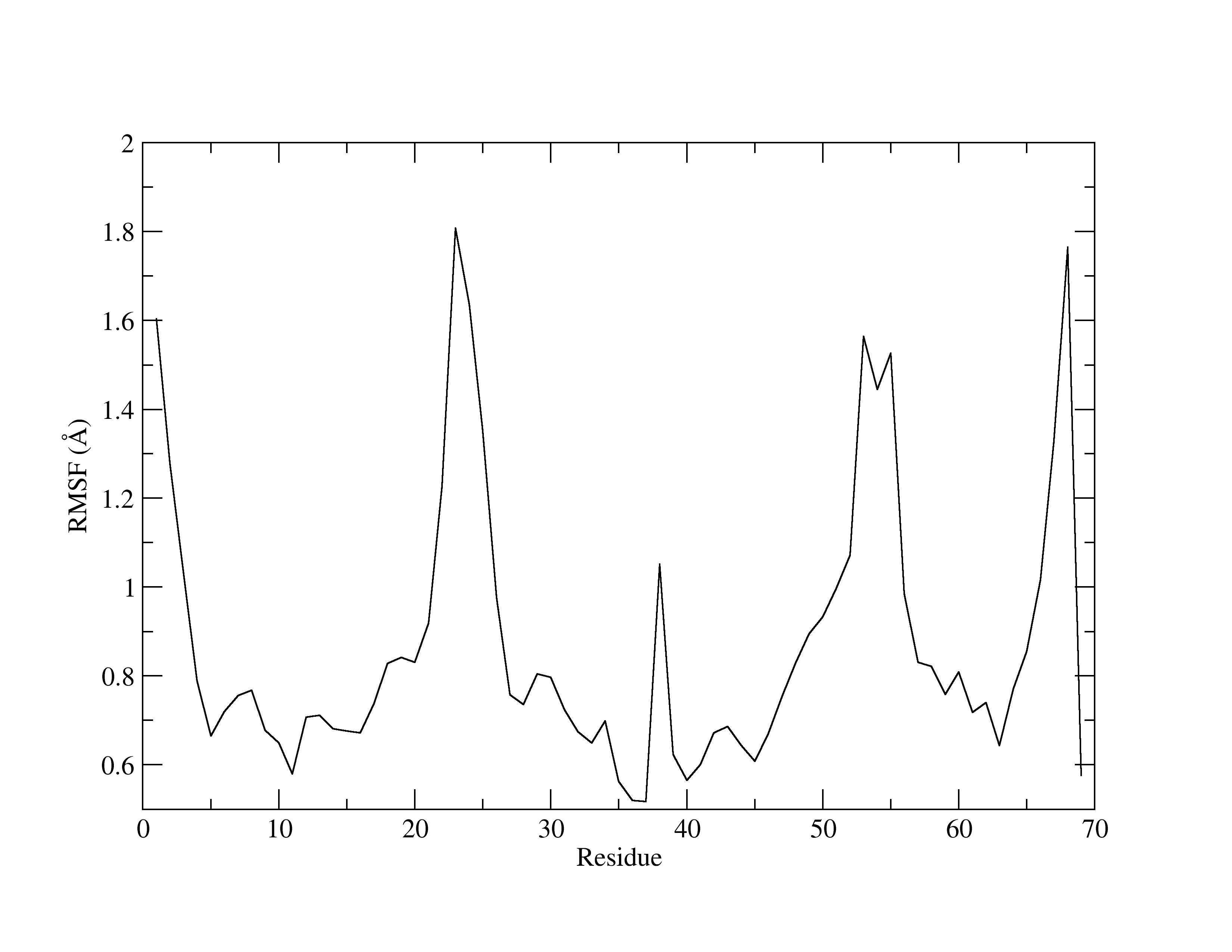
Figure 2. RMSF of MTR1.
The x-axis is the residue number, while the y-axis shows the fluctuations (in Å). Higher peaks indicate greater fluctuations. RMSFs are useful for identifying residues that have a greater contribution to the overall dynamics of the structure. Usually, it is common for residues involved in crystal contacts to exhibit higher peaks, reflecting increased mobility.
Computing Average Structures
Lastly, you will learn to obtain the average structure of a trajectory using CPPTRAJ with the command average.
Find the input file ctraj.average_MTR1.in and VIM it.
parm MTR1_prot-run.parm7
trajin centered.MTR1_prot-run.nc
average avg.pdb :1-69&!@H= pdb
run
quit
After the average command, you must provide a filename in which to save the data. Here, the name is avg.pdb, followed by the atom selection mask and the PDB file type specification pdb.
Run the input file as usual:
cpptraj -i ctraj.average_MTR1.in
Now, you have an average structure of MTR1 in a PDB format that can be opened up in VMD for viewing.