Divalent metal ion 12-6-4 models with Panteva–Giambasu–York (PGY) pairwise corrections for balanced interactions with nucleic acids
Learning objectives
Why do we need a good description of ions–nucleic acids interactions.
Know how to apply 12-6-4 parameters and the PGY pairwise corrections in Amber.
Relevant literature
Panteva, M., Giambasu, G., and York, D. M. Force Field for Mg2+, Mn2+, Zn2+, and Cd2+ Ions That Have Balanced Interactions with Nucleic Acids. J. Phys. Chem. B (2015). https://doi.org/10.1021/acs.jpcb.5b10423
Li, P., and Merz, K. M., Jr. Taking into Account the Ion-Induced Dipole Interaction in the Nonbonded Model of Ions. J. Chem. Theory Comput. (2013). https://doi.org/10.1021/ct400751u
Introduction
The need to model ions–nucleic acids interactions
Experimental and theoretical studies have shown that metal ions interact with nucleic acids in two main ways. First, electrostatic interactions between the negatively charged nucleic acid backbone and cations lead to a local enrichment of ions around the molecule, forming what is known as the ionic atmosphere. Its composition can be probed experimentally (for example by ion counting) and can also be described theoretically. Beyond these non-specific interactions, studies have also shown that both monovalent and divalent ions bind in a more specific manner, with well-localized binding sites. In RNA, these include coordination sites for divalent ions, especially Mg²⁺, whose presence can modify local geometry, stabilize compact tertiary folds, and interact with residues or ligands. These ions can also shift the pKa of nearby functional groups and contribute directly to catalytic activity. Such binding sites can be identified in X-ray crystallography and can also be investigated using NMR or spectroscopic techniques. However, all of these experimental approaches have limitations. In X-ray crystallography, for instance, it is often difficult to identify ions unambiguously; high, non-physiological ion concentrations are frequently required; and ion positions can be influenced by packing artifacts. As a result, many questions remain about the modes, locations, and strengths of ion binding to nucleic acids, as well as their impact on nucleic acid structure and dynamics. Molecular dynamics simulations therefore offer a valuable way to obtain a detailed, molecular-level description of these interactions, provided that they are able to represent them accurately…
Limitations in modelling ions–nucleic interactions
The main challenge in simulating divalent ions comes from the absence of electronic polarization in classical, non-polarizable force fields. Atomic charges are fixed and cannot adapt to the local electrostatic environment, even though electronic density redistribution is essential for describing ion–RNA interactions. This leads to several systematic artifacts: over-stabilization of interactions with non-bridging phosphate oxygens, under-stabilization of nucleobase interactions and an incorrect balance between solvation and direct coordination. As a consequence, divalent ions such as Mg²⁺ may accumulate unrealistically at the RNA surface and remain bound too strongly and the preferred binding sites can be poorly defined, with incorrect partners being favored. Similar, although weaker, effects can also be observed for monovalent ions and for interactions between charged residues, such as salt bridges.
How to overcome these limitations?
One option is to use polarizable force fields, which introduce additional terms into the standard functional form to account for electronic polarization explicitly. Approaches based on induced point dipoles and explicit multipoles provide a more realistic physical description of ion–RNA interactions. However, their higher computational cost and the difficulty of parameterization make them challenging to apply routinely, and they are not yet the preferred choice in large-scale biomolecular simulations.
An alternative is to modify the standard potential to incorporate key polarization effects implicitly. Several models follow this strategy by adjusting Lennard–Jones parameters or adding correction terms. The 12-6-4 model extends the classical Lennard–Jones potential by adding a charge-induced dipole interaction term in \(r^{-4}\):
The coefficients \(A_{ij}\) and \(B_{ij}\) correspond to the usual Lennard–Jones parameters, related to \(R_{ij}\) which is the distance at which two particles have a minimum in the LJ potential, while \(\varepsilon_{ij}\) is the well depth :
The additional coefficient \(C_{ij}\) introduces the charge-induced dipole interaction, which is defined as:
where \(\kappa\) is a scaling parameter (in units of Å⁻²) that determines the strength of the \(r^{-4}\) contribution.
However, although the original 12–6–4 model was developed to accurately reproduce bulk hydration properties, it fails to capture experimentally measured affinities for key RNA-binding sites. This limitation motivated the development of the m12–6–4 model, in which only the \(C_{ij}\) coefficients are re-optimized based on experimental data for three representative sites:
non-bridging oxygens of phosphate
N7 of adenine
N7 of guanine
The pairwise coefficient \(C_{ij}\) for an ion interacting with a given atom type is irectly proportional to the polarizability of the atom interacting with the ion \(\alpha\) (in \(\mathrm{Å^3}\)):
This yields several improvements:
more accurate binding free energies between X²⁺-phosphate groups
correct stabilization of nucleobase interactions
an improved balance between solvation and direct coordination
more realistic X²⁺–RNA dissociation kinetics
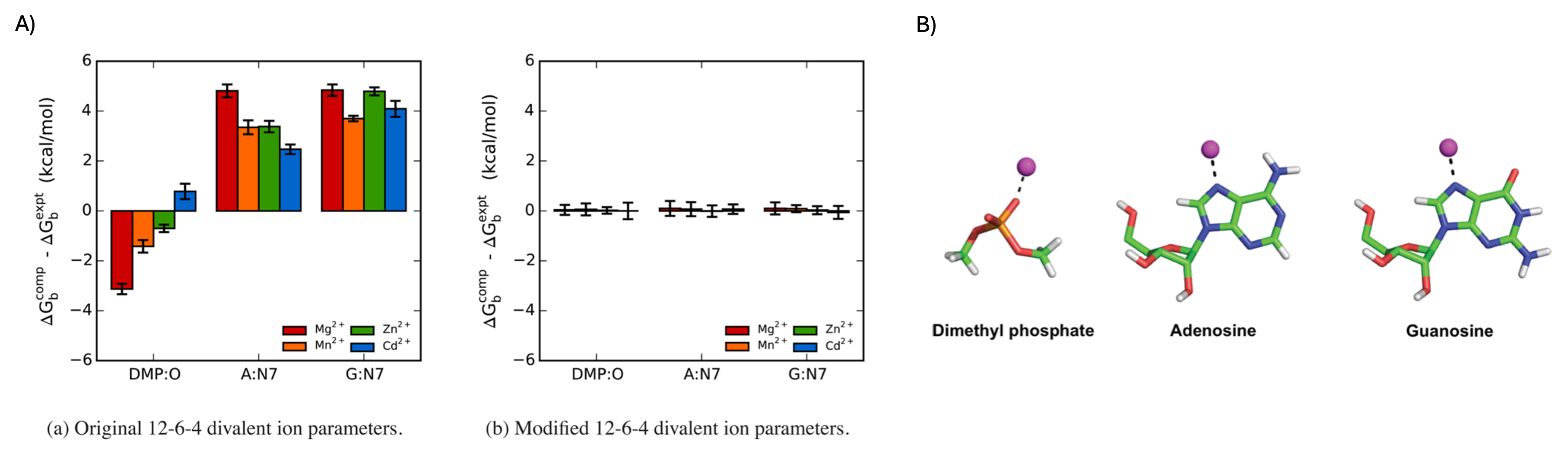
Figure 1. A) Comparison of the errors in the absolute binding free energies for the 12–6–4 (left) and m12–6–4 (right) Mg²⁺, Mn²⁺, Zn²⁺, and Cd²⁺ ion parameters for selected nucleic acid sites. Computed values are averages from three independent simulations, and the error bars correspond to the standard deviations. DMP = dimethyl phosphate, A = adenosine, G = guanosine. B) Model systems and binding sites for which pairwise parameters were tuned to reproduce the reference experimental binding free energies. The magenta sphere represents either Mg²⁺, Mn²⁺, Zn²⁺, or Cd²⁺.
Tutoriel
Downloadable files
The following files are required to build the 12-6-4 topology and apply the PGY (m12-6-4) pairwise corrections in Amber. They can be downloaded directly:
tleap-prep-PDB-2-Amber.intleap input file used to generate the initial Amber topology with the 12-6-4 ion model.1264.shShell script that prepares the ParmEd instruction file and applies the PGY corrections.parmed_1264_na.inParmEd instruction file assigning NAMG/NGMG/OPMG atom types and enabling the PGY corrections.lj_1264_pol.datPolarizability table used by add12_6_4 to compute the \(C_{ij}\) coefficients.
Building the topology with 12-6-4 parameters
In this activity, the goal is to learn how to apply these corrections for divalent cations interacting with nucleic acids. Magnesium will be used as an example here. The first step is to prepare a standard Amber topology using the 12-6-4 Mg²⁺ model. We assume you have an AmberTools environment activated, for example:
mamba activate AmberTools25
We start from an input PDB file named SYSTEM.pdb that contains an RNA molecule
and one or several Mg²⁺ ions.
Run tleap with the following input file.
The tleap input file
set default nocenter on # Do not recenter the molecule
### Sourcing amber force fields ###############################################
source leaprc.RNA.OL3 # RNA force field OL3
source leaprc.water.tip4pew # TIP4P-Ew water model
source leaprc.gaff2 # GAFF2 for small molecules
loadamberparams frcmod.ions234lm_1264_tip4pew # 12-6-4 metal-ion parameters
### Loading structure from file ##############################################
mol = loadpdb "SYSTEM.pdb"
### Adding counter ions ######################################################
addions mol NA 0 # Neutralize system
### Defining box & solvation #################################################
solvateoct mol TIP4PEWBOX 9 # 9 Å truncated octahedron box
### Adding bulk salt (example: 0.14 M NaCl) #################################
addionsrand mol NA 29 CL 29 6.0 # Random replacement, 6 Å min distance
### Saving ###################################################################
saveamberparm mol SYSTEM.parm7 SYSTEM.rst7
quit
After running:
tleap -f tleap-prep-PDB-2-Amber.in
you will obtain:
SYSTEM.parm7(topology)SYSTEM.rst7(coordinates)
These files include the 12-6-4 Mg²⁺ model.
Applying the PGY pairwise corrections
The m12-6-4 (PGY) corrections are applied in a second pass using ParmEd.
The script 1264.sh
This helper script inserts the proper output file names and runs ParmEd:
#!/bin/bash
# Usage: ./1264.sh SYSTEM
name=$1
cat parmed_1264_na.in > parmed.${name}.in
sed -i '/^outparm/d' parmed.${name}.in
cat <<EOF >> parmed.${name}.in
outparm ${name}_1264.parm7 ${name}_1264.rst7
EOF
parmed -i parmed.${name}.in -p ${name}.parm7 -c ${name}.rst7
Make executable:
chmod +x 1264.sh
The file parmed_1264_na.in
This file applies the PGY-specific N7(A), N7(G), and OP corrections:
setOverwrite True
change AMBER_ATOM_TYPE :A*,DA*@N7 NAMG
change AMBER_ATOM_TYPE :G*,DG*@N7 NGMG
change AMBER_ATOM_TYPE :*@OP* OPMG
addLJType @%NAMG
addLJType @%NGMG
addLJType @%OPMG
add12_6_4 @%Mg2+ watermodel TIP4PEW polfile lj_1264_pol.dat
printLJMatrix @%Mg2+
outparm NAME.parm7 NAME.rst7
Polarizabilities (lj_1264_pol.dat)
Below is the full content of the lj_1264_pol.dat file:
H 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HC 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H0 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H1 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H2 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H3 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HA 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H4 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H5 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HO 0.000 Keep its C4 terms consistent with its 12-6 LJ parameters
HS 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HP 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HZ 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h1 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h2 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h3 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h4 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h5 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
ha 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
hc 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
hn 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
ho 0.000 Keep its C4 terms consistent with its 12-6 LJ parameters
hp 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
hs 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
hx 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
Hc 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
Ha 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
Ho 0.000 Keep its C4 terms consistent with its 12-6 LJ parameters
Hp 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
C 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C* 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C4 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C5 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CA 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CB 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CC 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CD 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CK 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CM 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CN 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CO 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CP 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CQ 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CR 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CS 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CV 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CW 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CY 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CZ 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C2 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C1 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
c 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
c2 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
ca 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cc 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cd 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
ce 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cf 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cp 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cq 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cu 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cv 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cx 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cy 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cz 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
2C 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
3C 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
C8 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
CI 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
CT 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
CX 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
XC 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
TG 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
c3 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
C7 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
CJ 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
c1 1.283 Referenced or adopted from Miller JACS,112,8533(1990)
cg 1.283 Referenced or adopted from Miller JACS,112,8533(1990)
ch 1.283 Referenced or adopted from Miller JACS,112,8533(1990)
Cg 1.061 Referenced or adoptedfrom Miller JACS,112,8533(1990)
Cy 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
Cp 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
Ck 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
Cj 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
N 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
N* 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
N2 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
N3 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NA 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NB 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NAMG 1.910 From Panteva et al. JPCB, 2015,119,15460
NGMG 1.925 From Panteva et al. JPCB, 2015,119,15460
NAMN 1.770 From Panteva et al. JPCB, 2015,119,15460
NGMN 1.860 From Panteva et al. JPCB, 2015,119,15460
NAZN 1.480 From Panteva et al. JPCB, 2015,119,15460
NGZN 1.640 From Panteva et al. JPCB, 2015,119,15460
NACD 1.580 From Panteva et al. JPCB, 2015,119,15460
NGCD 1.920 From Panteva et al. JPCB, 2015,119,15460
NC 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
ND 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NL 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NT 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NY 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
TN 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n1 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n2 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n3 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n4 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
na 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nb 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nc 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nd 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
ne 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nf 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nh 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
no 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
Ng 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
O 0.569 Referenced or adopted from Miller JACS,112,8533(1990)
O2 0.569 Referenced or adopted from Miller JACS,112,8533(1990)
O3 0.569 Referenced or adopted from Miller JACS,112,8533(1990)
OD 0.569 Referenced or adoptedfrom Miller JACS,112,8533(1990)
OP 0.569 Referenced or adopted from Miller JACS,112,8533(1990)
OPMG 0.170 From Panteva et al. JPCB, 2015,119,15460
OPMN 0.370 From Panteva et al. JPCB, 2015,119,15460
OPZN 0.510 From Panteva et al. JPCB, 2015,119,15460
OPCD 0.680 From Panteva et al. JPCB, 2015,119,15460
OA 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
OH 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
OS 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
OY 0.637 Added by AG
OZ 0.637 Added by AG
OX 0.637 Added by AG
OW 1.444 From "The Structure and Properties of Water" by Eisenberg & Kauzmann
o 0.569 Referenced or adoptedfrom Miller JACS,112,8533(1990)
oh 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
os 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
ow 1.444 From "The Structure and Properties of Water" by Eisenberg & Kauzmann
Oh 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
Os 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
Oy 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
S 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
SH 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
s 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
s2 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
s4 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
s6 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
sh 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
ss 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
sx 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
sy 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
Sm 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
P 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
p2 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
p3 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
p4 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
p5 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pb 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pc 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pd 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pe 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pf 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
px 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
py 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
PX 1.538 Added by AG
F 0.32 From Applequist et al. JACS,94,2952(1972)
Cl 1.91 From Applequist et al. JACS,94,2952(1972)
Br 2.88 From Applequist et al. JACS,94,2952(1972)
I 4.69 From Applequist et al. JACS,94,2952(1972)
f 0.32 From Applequist et al. JACS,94,2952(1972)
cl 1.91 From Applequist et al. JACS,94,2952(1972)
br 2.88 From Applequist et al. JACS,94,2952(1972)
i 4.69 From Applequist et al. JACS,94,2952(1972)
Li 0.029 From Sangster & Atwood J. Phys. C 11,1541(1978)
Li+ 0.029 From Sangster & Atwood J. Phys. C 11,1541(1978)
IP 0.2495 From Sangster & Atwood J. Phys. C 11,1541(1978)
Na 0.2495 From Sangster & Atwood J. Phys. C 11,1541(1978)
Na+ 0.2495 From Sangster & Atwood J. Phys. C 11,1541(1978)
K 1.0571 From Sangster & Atwood J. Phys. C 11,1541(1978)
K+ 1.0571 From Sangster & Atwood J. Phys. C 11,1541(1978)
Rb 1.5600 From Sangster & Atwood J. Phys. C 11,1541(1978)
Rb+ 1.5600 From Sangster & Atwood J. Phys. C 11,1541(1978)
Cs 2.5880 From Sangster & Atwood J. Phys. C 11,1541(1978)
Cs+ 2.5880 From Sangster & Atwood J. Phys. C 11,1541(1978)
F- 0.9743 From Sangster & Atwood J. Phys. C 11,1541(1978)
Cl- 3.2350 From Sangster & Atwood J. Phys. C 11,1541(1978)
IM 3.2350 From Sangster & Atwood J. Phys. C 11,1541(1978)
Br- 4.5330 From Sangster & Atwood J. Phys. C 11,1541(1978)
I- 6.7629 From Sangster & Atwood J. Phys. C 11,1541(1978)
Be2+ 0.0067 Calculated from B3LYP/6-311++G(2d,2p)
Cu2+ 0.413 Calculated from B3LYP/6-311++G(2d,2p)
CU 0.413 Calculated from B3LYP/6-311++G(2d,2p)
Ni2+ 0.395 Calculated from B3LYP/6-311++G(2d,2p)
Zn 0.344 Calculated from B3LYP/6-311++G(2d,2p)
ZN 0.344 Calculated from B3LYP/6-311++G(2d,2p)
Zn2+ 0.344 Calculated from B3LYP/6-311++G(2d,2p)
Co2+ 0.447 Calculated from B3LYP/6-311++G(2d,2p)
Cr2+ 0.623 Calculated from B3LYP/6-311++G(2d,2p)
Fe 0.518 Calculated from B3LYP/6-311++G(2d,2p)
FE 0.518 Calculated from B3LYP/6-311++G(2d,2p)
Fe2+ 0.518 Calculated from B3LYP/6-311++G(2d,2p)
Mg 0.048 Calculated from B3LYP/6-311++G(2d,2p)
MG 0.048 Calculated from B3LYP/6-311++G(2d,2p)
Mg2+ 0.048 Calculated from B3LYP/6-311++G(2d,2p)
V2+ 0.620 Calculated from B3LYP/6-311++G(2d,2p)
Mn2+ 0.534 Calculated from B3LYP/6-311++G(2d,2p)
Hg2+ 0.707 Calculated from B3LYP/SDD
Cd2+ 0.427 Calculated from B3LYP/SDD
Ca2+ 0.477 Calculated from B3LYP/6-311++G(2d,2p)
C0 0.477 Calculated from B3LYP/6-311++G(2d,2p)
Sn2+ 3.083 Calculated from B3LYP/SDD
Sr2+ 0.813 Calculated from B3LYP/SDD
Ba2+ 1.496 Calculated from B3LYP/SDD
HW 0.000 Water hydrogen / dummy atom
hw 0.000 Water hydrogen / dummy atom
EP 0.000 Water hydrogen / dummy atom
EPW 0.000 Water hydrogen / dummy atom
EP1 0.000 Water hydrogen / dummy atom
EP2 0.000 Water hydrogen / dummy atom
LP 0.000 Lone pair site
Al3+ 0.031 Calculated from B3LYP/6-311++G(2d,2p)
Fe3+ 0.297 Calculated from B3LYP/6-311++G(2d,2p)
Cr3+ 0.352 Calculated from B3LYP/6-311++G(2d,2p)
Y3+ 0.597 Calculated from B3LYP/SDD
La3+ 1.141 Calculated from B3LYP/SDD
Pr3+ 1.101 Calculated from B3LYP/SDD
Nd3+ 1.047 Calculated from B3LYP/SDD
Sm3+ 0.927 Calculated from B3LYP/SDD
Eu3+ 0.885 Calculated from B3LYP/SDD
Tm3+ 0.675 Calculated from B3LYP/SDD
Lu3+ 0.629 Calculated from B3LYP/SDD
Zr4+ 0.436 Calculated from B3LYP/SDD
U4+ 1.044 Calculated from B3LYP/SDD
Th4+ 1.141 Calculated from B3LYP/SDD
M0 0.000 Treat as zero
Output files
Running:
./1264.sh SYSTEM
produces:
SYSTEM_1264.parm7SYSTEM_1264.rst7
Figure 2. Screenshot of the output showing the interaction matrix involving magnesium.