2. Hands-On Session 2: Perform molecular dynamics (MD) simulations using AMBER
2.1. Learning Objectives
Understand the basic workflow of molecular dynamics (MD) simulations using AMBER, from equilibration to production.
Perform stepwise equilibration, apply and gradually release positional restraints during equilibration.
Read and modify AMBER input files (.mdin) for common MD stages (minimization, heating, equilibration, production).
Use cpptraj to process MD outputs and prepare structures for visualization.
Visualize MD results with VMD and compare structures before and after equilibration.
Understand the special considerations made to stably equilibrating a nucleic acid system.
2.2. Activities
flowchart LR
%% =========================
%% Equilibration
%% =========================
A2["Input files<br/>MTR1_counterIon_water_bulk.parm7"]
A1["Input files<br/>MTR1_counterIon_water_bulk.rst7<br/>mdins/*.mdin<br/>restrnt.disang"]
P1{{"<b>[§2.3.4.2]</b><br/>Run equilibration<br/>sander / pmemd.cuda"}}
O1_1["<b>[from §2.3.4.2]</b><br/>Equilibration outputs<br/>mdout<br/>equil.nc<br/>"]
O1_2["<b>[from §2.3.4.2]</b><br/>Final restart<br/>09_Equil_solute_Rwt2-run.rst7"]
A1 --> P1
A2 --> P1
P1 --> O1_1
P1 --> O1_2
%% =========================
%% Wrap equilibrated restart and trajectory
%% =========================
P2{{"<b>[§2.3.4.3]</b><br/>Wrap equilibration<br/>cpptraj"}}
O2["<b>[from §2.3.4.3]</b><br/>Wrapped equilibration files<br/>centered.09_Equil_solute_Rwt2-run.rst7<br/>centered_equil.nc"]
O1_1 --> P2
O1_2 --> P2
P2 --> O2
%% =========================
%% Inspect equilibration
%% =========================
P3{{"<b>[§2.3.4.3]</b><br/>Inspect equilibration<br/>VMD"}}
O3["Equilibration inspection<br/>RNA RMSD<br/>ion positions"]
O2 --> P3
P3 --> O3
%% =========================
%% Prepare and Run Production
%% =========================
P4{{"<b>[§2.3.5.2]</b><br/>Apply HMR<br/>cpptraj"}}
O4["<b>[from §2.3.5.2]</b><br/>HMR topology<br/>MTR1_counterIon_water_bulk-run.parm7"]
A2 --> P4
P4 --> O4
P5{{"<b>[§2.3.5.3]</b><br/>Run production MD<br/>pmemd.cuda"}}
O5_1["<b>[from §2.3.5.3]</b><br/>Production logs<br/>MTR1_counterIon_water_bulk-run-01.mdout<br/>MTR1_counterIon_water_bulk-run-01.mdinfo"]
O5_2["<b>[from §2.3.5.3]</b><br/>Production trajectory/restart<br/>MTR1_counterIon_water_bulk-run-01.nc<br/>MTR1_counterIon_water_bulk-run-01.rst7"]
O1_2 --> P5
O4 --> P5
P5 --> O5_1
P5 --> O5_2
P6{{"<b>[§2.3.5.4]</b><br/>Wrap production<br/>cpptraj"}}
O6["<b>[from §2.3.5.4]</b><br/>Wrapped production files<br/>centered.MTR1_counterIon_water_bulk-run-01.nc<br/>centered.MTR1_counterIon_water_bulk-run-01.rst7"]
O5_2 --> P6
P6 --> O6
O7["Production inspection<br/>RNA RMSD<br/>ion positions"]
P7{{"<b>[§2.3.5.4]</b><br/>Inspect production<br/>VMD"}}
O6 --> P7
P7 --> O7
%% =========================
%% Styling
%% =========================
classDef file fill:#fff7e6,stroke:#d98c00,stroke-width:1.5px,color:#111;
classDef program fill:#e8f1ff,stroke:#1f77b4,stroke-width:1.8px,color:#111;
classDef result fill:#eaf7ea,stroke:#2ca02c,stroke-width:1.5px,color:#111;
class A1,A2 file;
class P1,P2,P3,P4,P5,P6,P7 program;
class O1_1,O1_2,O2,O3,O4,O5_1,O5_2,O6,O7 result;
2.2.1. Starting structure and input files
This activity starts from pre-generated AMBER topology and coordinate files that define the initial structure of the fully solvated MTR1 ribozyme system [1][2] with counterions, described using the OL3 RNA force field [3] and the TIP4P-Ew water model [4]. These files serve as the starting point for all subsequent equilibration and production molecular dynamics simulations performed in this activity.
Starting from these files, the system will be equilibrated stepwise and then used to run production molecular dynamics simulations, generating new restart and trajectory files at each stage.
The specific starting files used in this activity are MTR1_counterIon_water_bulk.parm7 and MTR1_counterIon_water_bulk.rst7.
These files are used to demonstrate the complete equilibration and production molecular dynamics workflow.
2.2.2. Accessing the activity files
All input files and reference output files required for this activity are available on the DIPC cluster and as a downloadable archive.
DataDir="/data/amber26/TutorialData/HandsOn2_MD"
TutorialDir="/scratch/$USER/TutorialData/HandsOn2_MD"
mkdir -p $TutorialDir
cp -r $DataDir/* $TutorialDir
cd $TutorialDir
cp -r input working_dir
cd working_dir
Option B — Downloading the archive
Hands-On Session 2 files archive (tar.gz)
tar -xzf HandsOn2_MD.tar.gz
cd HandsOn2_MD
2.2.3. Equilibration and Production Workflow
flowchart LR
A["Initial system<br/>Inputs: parm7, rst7 (ions and solvent)"]
subgraph EQ["Equilibration"]
direction LR
B["Solvent equilibration<br/>Restrained solute"]
C["Heating to 300 K"]
D["Gradual solute release"]
E["Wrap and inspect equilibration<br/>Centered equilibration structure"]
B --> C --> D --> E
end
subgraph PR["Production"]
direction LR
F["Production MD<br/>Unrestrained system"]
G["Wrap and inspect production<br/>Centered production trajectory"]
F --> G
end
subgraph AN["Analysis"]
direction LR
H["Final inspection and analysis<br/>RMSD, visualization"]
end
A --> B
E --> F
G --> H
2.2.4. Equilibrating the Box
Change into the Equil directory to run the stepwise equilibration of the system.
cd Equil
Now you will learn how to equilibrate the simulation box. Electrostatic interactions are extremely important for RNA structure given the negatively charged backbone. Therefore, a rigorous equilibration process allows the solvent to properly organize around the molecule in a controlled way while avoiding large fluctuations in density that could cause errors. This is especially important when your system contains divalent metal ions. The electrostatic interaction energy is proportional to the product of the two charges, so increasing the charge will lead to a larger interaction energy at a given distance. Nucleic acids are highly charged systems and must be equilibrated slowly. The equilibration procedure used here follows the protocol described in [5].
The Equil directory contains
eqFromStratch.slurm and a directory called mdins.
List the contents of the mdins directory:
ls mdins
000_Min.mdin 003_NPT.mdin 03_Equil_NPT_solvent.mdin 06_Equil_solute_Rwt25.mdin 09_Equil_solute_Rwt2.mdin
001_NPT.mdin 01_Min.mdin 04_Min_solute.mdin 07_Equil_solute_Rwt10.mdin
002_NPT.mdin 02_Heat.mdin 05_Heat_solute_Rwt25.mdin 08_Equil_solute_Rwt5.mdin
These are the input files for the stepwise equilibration procedure and subsequent production simulation.
Note
The general approach is to slowly equilibrate the solvent around the solute, then gradually allow the solute to move. We cycle through minimizations to remove clashes and find a local minimum, heating to explore new conformations, then simulations in the NPT ensemble to approach the global minimum. There are some specific points to note about how the provided mdin files have been written.
2.2.4.1. Equilibration input files
Initial energy minimization of the solvent. Solute atoms are positionally restrained with weights of 50 kcal/mol Ų.
The line highlighted in the initial minimization input file specifies the positional restraint mask used during equilibration. Restraints are applied to all atoms that are not hydrogens and not part of the solvent or ions (
NA,CL,MG,WAT). As a result, all heavy atoms of the solute are restrained, while solvent molecules and ions are free to relax. In practice, this effectively keeps the RNA heavy atoms close to their starting positions during the early stages of equilibration. There is no Mg2+ in this system currently, but many RNA systems will contain it. If additional solvent components are present, they should be included in the restraint mask. When ions were added in LEaP they were referred to asNAandCL, but equivalent residue names such asNa+andCl-would also be handled correctly by the mask.Another highlighted line in the same input file indicates that a file named
restrnt.disangis read as input. This file can contain NMR-style restraints, such as distance, angle, or torsion restraints between selected atoms. While coordinate restraints work with respect to another structure file, NMR restraints (enabled byntr = 1) work with respect to other atoms within the same structure. This way atoms are allowed to fluctuate while maintaining defined geometric relationships.
000_Min.mdin:
Initial Solvent Energy Minimization Stage
Bulk water and Sodium ions only
&cntrl
imin = 1
ntmin = 0
maxcyc = 500
ntx = 1
ntpr = 250
ntwx = 0
ntr = 1
restraint_wt = 50.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
&wt type = 'DUMPFREQ', istep1 = 10000, /
&wt type='END' /
DISANG=restrnt.disang
DUMPAVE=restrnt.dumpave
LISTOUT=POUT
LISTIN=POUT
Initial equilibration in the NPT ensemble. Temperature is held at 300 K and pressure at 1 atm for 5 ps. Solute atoms are positionally restrained with weights of 50 kcal/mol Ų.
001_NPT.mdin:
&cntrl
imin = 0
ntx = 1
nstlim = 2500
ntb = 2
ntp = 1
temp0 = 300.0
ntr = 1
restraint_wt = 50.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
Additional equilibration in the NPT ensemble. Identical to the previous step, but velocities are read to allow gradual box relaxation and avoid large density fluctuations.
002_NPT.mdin:
&cntrl
imin = 0
ntx = 5
nstlim = 2500
ntb = 2
ntp = 1
temp0 = 300.0
ntr = 1
restraint_wt = 50.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
Extended equilibration in the NPT ensemble. This step is run continuously for 190 ps to complete a total of 200 ps of NPT equilibration.
003_NPT.mdin:
&cntrl
imin = 0
ntx = 5
nstlim = 95000
ntb = 2
ntp = 1
temp0 = 300.0
ntr = 1
restraint_wt = 50.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
Solvent energy minimization. Solute atoms remain positionally restrained to remove residual clashes after initial equilibration.
01_Min.mdin:
&ntrl
imin = 1
ntmin = 0
maxcyc = 500
ntx = 1
ntpr = 250
ntwx = 0
ntr = 1
restraint_wt = 50.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
DISANG=restrnt.disang
Initial heating at constant volume. The temperature is increased from 0 to 300 K over 600 ps and then held constant for 1 ns, with positional restraints applied.
02_Heat.mdin:
&cntrl
imin = 0
ntx = 1
nstlim = 800000
dt = 0.002
ntc = 2
ntf = 2
ntb = 1
ntt = 3
tempi = 0.0
temp0 = 300.0
gamma_ln = 2.0
ntpr = 500
ntwx = 500
ntr = 1
restraint_wt = 50.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
Solvent equilibration in the NPT ensemble. Temperature is held at 300 K and pressure at 1 atm for 5 ns while the solute remains restrained.
03_Equil_NPT_solvent.mdin:
&cntrl
imin = 0
ntx = 5
nstlim = 2500000
dt = 0.002
ntc = 2
ntf = 2
ntb = 2
ntp = 1
pres0 = 1.0
taup = 1.0
ntt = 3
temp0 = 300.0
gamma_ln = 2.0
ntpr = 5000
ntwx = 5000
ntr = 1
restraint_wt = 50.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
Solute energy minimization. Positional restraints are released to relax the solute and solvent with respect to each other.
04_Min_solute.mdin:
&cntrl
imin = 1
ntmin = 0
maxcyc = 500
ntx = 1
ntpr = 250
ntwx = 0
ntr = 0
cut = 12.0
/
Solute heating with reduced restraints. Increase the temperature from 0 to 300 K over 600 ps, then hold at constant temperature for 1 ns. Solute atoms are positionally restrained with weights of 25 kcal/mol². The restraints are released gradually over ensuing stages of dynamics. Changing restraints effectively changes the force field. If the energy associated with a restraint is high and it is suddenly removed, the bonds to the associated atom could be broken as the atom position changes dramatically in a step of dynamics.
05_Heat_solute_Rwt25.mdin:
&cntrl
imin = 0
ntx = 1
nstlim = 800000
dt = 0.002
ntc = 2
ntf = 2
ntb = 1
ntt = 3
tempi = 0.0
temp0 = 300.0
gamma_ln = 2.0
ntpr = 500
ntwx = 500
ntr = 1
restraint_wt = 25.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
Solute equilibration with 25 kcal/mol Ų restraints. Short 500 ps NPT equilibration at reduced restraint strength.
06_Equil_solute_Rwt25.mdin:
&cntrl
imin = 0
ntx = 5
nstlim = 250000
dt = 0.002
ntc = 2
ntf = 2
ntb = 2
ntp = 1
pres0 = 1.0
taup = 1.0
ntt = 3
temp0 = 300.0
gamma_ln = 2.0
ntpr = 5000
ntwx = 5000
ntr = 1
restraint_wt = 25.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
Solute equilibration with 10 kcal/mol Ų restraints. During 500 ps.
07_Equil_solute_Rwt10.mdin:
&cntrl
imin = 0
ntx = 5
nstlim = 250000
dt = 0.002
ntc = 2
ntf = 2
ntb = 2
ntp = 1
pres0 = 1.0
taup = 1.0
ntt = 3
temp0 = 300.0
gamma_ln = 2.0
ntpr = 5000
ntwx = 5000
ntr = 1
restraint_wt = 10.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
Solute equilibration with 5 kcal/mol Ų restraints. During 500 ps.
08_Equil_solute_Rwt5.mdin:
&cntrl
imin = 0
ntx = 5
nstlim = 250000
dt = 0.002
ntc = 2
ntf = 2
ntb = 2
ntp = 1
pres0 = 1.0
taup = 1.0
ntt = 3
temp0 = 300.0
gamma_ln = 2.0
ntpr = 5000
ntwx = 5000
ntr = 1
restraint_wt = 5.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
Final solute equilibration with weak restraints. Positional restraints are reduced to 2 kcal/mol Ų during 500 ps, prior to production MD.
09_Equil_solute_Rwt2.mdin:
&cntrl
imin = 0
ntx = 5
nstlim = 250000
dt = 0.002
ntc = 2
ntf = 2
ntb = 2
ntp = 1
pres0 = 1.0
taup = 1.0
ntt = 3
temp0 = 300.0
gamma_ln = 2.0
ntpr = 5000
ntwx = 5000
ntr = 1
restraint_wt = 2.0
restraintmask = '!@H= & !:NA,CL,MG,WAT'
cut = 12.0
/
In this activity, no NMR restraints are applied; however, this line is retained so that a single, centralized set of MD input files can be used whether or not additional restraints are required. In this case,``restrnt.disang`` is intentionally empty.
Create an empty restraint file in the Equil directory:
touch restrnt.disang
2.2.4.2. Running the equilibration
You have been provided the SLURM submission script
eqFromStratch.slurm, which automates the full stepwise equilibration
procedure described above.
Take a look at the contents of eqFromStratch.slurm:
#!/bin/bash
#SBATCH --qos=regular
#SBATCH --time=1-00:00:00
#SBATCH --job-name=equil
#SBATCH --reservation=amber26
#SBATCH --nodes=1
#SBATCH --gpus=1
#SBATCH --ntasks-per-node=1
#SBATCH --mem=10G
#SBATCH --output=equil-%j.out
#SBATCH --error=equil-%j.err
module load Amber/25.0-foss-2022a-AmberTools-25-CUDA-12.1.1-DeePMD
set -x
echo AMBERHOME is $AMBERHOME
test -f ${AMBERHOME}/amber.sh && source ${AMBERHOME}/amber.sh
LAUNCH="srun --kill-on-bad-exit"
EXE=${AMBERHOME}/bin/pmemd.cuda
BASE=$1
PARM=${BASE}.parm7
REF=${BASE}.rst7
mdins=mdins
NAME=${BASE}
steps="\
000_Min \
001_NPT \
002_NPT \
003_NPT \
01_Min \
02_Heat \
03_Equil_NPT_solvent \
04_Min_solute \
05_Heat_solute_Rwt25 \
06_Equil_solute_Rwt25 \
07_Equil_solute_Rwt10 \
08_Equil_solute_Rwt5 \
09_Equil_solute_Rwt2 \
"
for step in $steps; do
ICRD=${BASE}.rst7
MDIN=${mdins}/${step}.mdin
BASE=${step}-run
# The first two steps are minimizations, which run on the CPU with sander.
# Every other step runs on the GPU with pmemd.cuda.
if [ "$step" == 000_Min ] || [ "$step" == 01_Min ]; then
restartfile=${BASE}.rst7
trajFile=${BASE}.rst7
EXE=${AMBERHOME}/bin/sander
else
restartfile=${BASE}.rst7
trajFile=${BASE}.nc
EXE=${AMBERHOME}/bin/pmemd.cuda
fi
echo running ${MDIN}
${LAUNCH} ${EXE} -O -p ${PARM} -c ${ICRD} -i ${MDIN} -o ${BASE}.mdout -inf ${BASE}.mdinfo -x ${BASE}.nc -r ${BASE}.rst7 -ref ${REF}
mv ${BASE}.mdinfo ${BASE}.stat
done
This script will take as input the prefix of the parameter and restart
files. The equilibration steps are run one at a time. Highlighted
lines indicate whether early minimization steps are performed with
sander or whether the simulation is run with pmemd.cuda. This is
because pmemd.cuda is more sensitive to large spikes in the energy
than sander, and we want to avoid errors.
The equilibration script takes a single argument, referred to as BASE,
which defines the prefix of the AMBER topology and coordinate files used as
input.
In this activity, BASE is set to MTR1_counterIon_water_bulk.
The script therefore expects the following files to be present in the working directory:
These files are used as the starting point for the first equilibration step, and subsequent steps will automatically use the restart file generated by the previous step.
Warning
However, the duration of the equilibration procedure is too long for this workshop; but the job would be submitted as follows:
sbatch eqFromStratch.slurm MTR1_counterIon_water_bulk
2.2.4.3. Wrapping and inspection
The outputs of the equilibration procedure have been provided for you in
the output/Equil directory. Now we will compare the structure
from before and after equilibration. The last step of equilibration
produced output with the prefix 09_Equil_solute_Rwt2-run. Now that we
have performed simulations under periodic boundary conditions, atoms may
have diffused into neighboring images. To visualize a single periodic
unit, we need to wrap the solvent around the RNA, which is a concept you
have been introduced to in a previous session. You have been provided a
cpptraj input file called wrap_RNA_equil.in.
Copy the outputs of the equilibration to your working directory:
cp ../../output/Equil/0*-run* ./
The wrap_RNA_equil.in input file contains:
parm MTR1_counterIon_water_bulk.parm7
trajin 09_Equil_solute_Rwt2-run.rst7
reference 09_Equil_solute_Rwt2-run.rst7
center '!@H= & !:MG= & !:NA= & !:CL= & !:WAT=' mass origin
autoimage '!@H= & !:MG= & !:NA= & !:CL= & !:WAT='
center '!@H= & !:MG= & !:NA= & !:CL= & !:WAT=' mass reference
trajout centered.09_Equil_solute_Rwt2-run.rst7
run
quit
In this input file we set the center of mass of the RNA at the origin,
then we image the solute around the RNA. Finally, we center the imaged
system based on the reference restart file and output a new restart file
with the prefix centered to distinguish it. Now we can inspect the
wrapped structure in VMD.
Wrap the structure 09_Equil_solute_Rwt2-run.rst7 (the last frame output during equilibration):
module load Amber/25.0-foss-2022a-AmberTools-25-CUDA-12.1.1-DeePMD
cpptraj -i wrap_RNA_equil.in
Open the structure before and after equilibration in VMD locally. First, download the necessary files:
rsync -avz "username@hyperion.sw.ehu.es:/scratch/amber26-XX/TutorialData/HandsOn2_MD/working_dir/Equil/{centered.09_Equil_solute_Rwt2-run.rst7,MTR1_counterIon_water_bulk.rst7,MTR1_counterIon_water_bulk.parm7}" .
Locally: Open the structure before and after equilibration in VMD:
vmd -f MTR1_counterIon_water_bulk.parm7 \
centered.09_Equil_solute_Rwt2-run.rst7 \
-f MTR1_counterIon_water_bulk.parm7 \
MTR1_counterIon_water_bulk.rst7
For each selected molecule, change the representation from all to
all nucleic.
Using the RMSD Calculator tool, uncheck the Backbone only box if it is
checked, align the structures with the selection nucleic and noh and
calculate the RMSD.
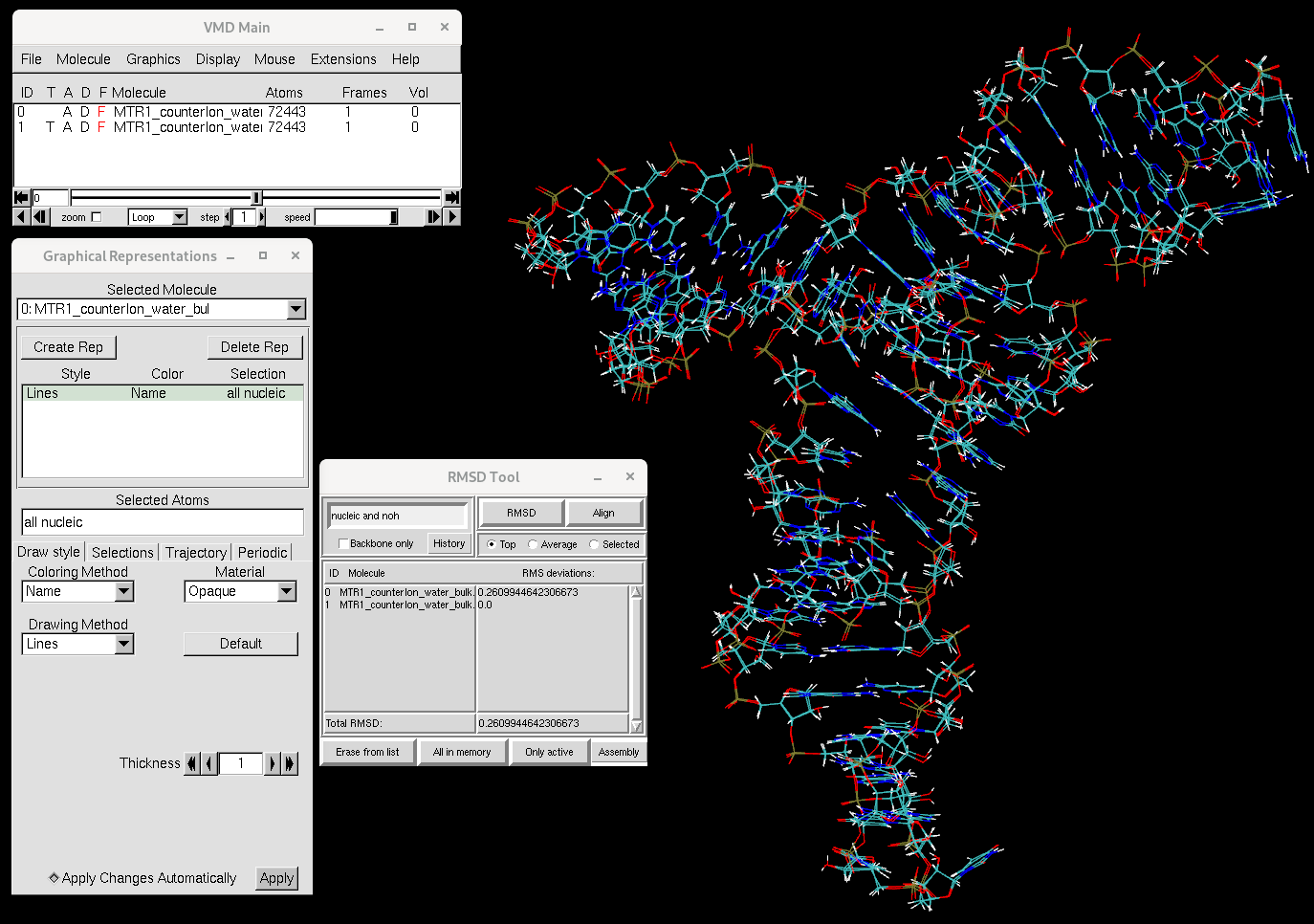
Figure 1. RMSD between the MTR1 structure before and after equilibration. RMSD is computed for RNA atoms.
You will see that the RNA structure has not changed much over the course of equilibration. Now let’s focus on the ions.
In the RMSD Calculator, change the selection from nucleic and noh to
ions.
Uncheck the Backbone only option if it is checked and compute the RMSD for the ions.
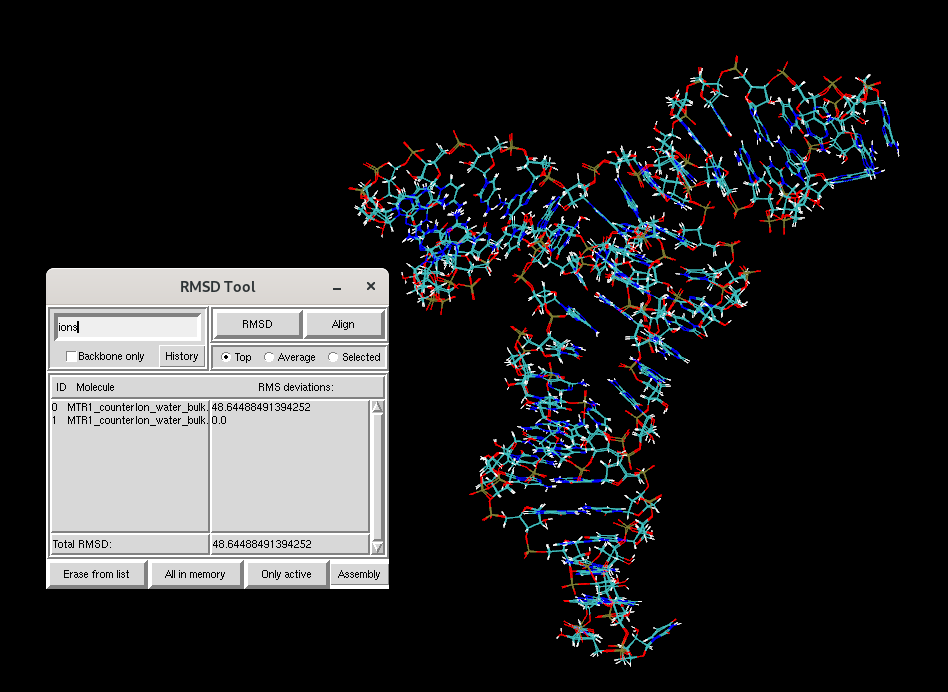
Figure 2. RMSD of ions in the MTR1 simulation box before and after equilibration.
You will see that the RMSD for the ions between the two structures is much larger than that of the RNA. To study the dynamics of the RNA, you will next learn to run production MD simulations starting from your equilibrated structure.
2.2.5. Running Production Simulations
In this section, you will run a very short 0.5 ns production molecular dynamics simulation without positional restraints on the RNA. In previous studies of this RNA system, production simulations converged on the order of tens of nanoseconds. Although longer time scales are desirable, such simulations cannot be performed within the time constraints of this workshop.
2.2.5.1. Preparing the production input files
Create a new working directory for production and copy all necessary files:
cd ../Prod
cp ../Equil/MTR_waterreps.txt ./
The production MD input file 11_Production_0.5ns.mdin performs
0.5 ns of unrestrained dynamics in the isothermal–isobaric ensemble (NPT)
and is intended to provide a brief introduction to running production
simulations in AMBER.
Production MD will be run with a 4 fs timestep. To do this stably, hydrogen
mass repartitioning (HMR) is applied. The cpptraj input file
hmr.in used to apply hydrogen mass repartitioning is shown below:
parm MTR1_counterIon_water_bulk.parm7
hmassrepartition
parmwrite out MTR1_counterIon_water_bulk-run.parm7
go
quit
Run cpptraj to generate the HMR-modified topology file:
cpptraj -i hmr.in
This command generates MTR1_counterIon_water_bulk-run.parm7, which will be used for the production simulation.
Create an empty restraint file in the production directory:
touch restrnt.disang
2.2.5.2. The production SLURM script
The following SLURM script prod.slurm is used to run the production
simulation. It takes as input the prefix of the AMBER topology and
restart files and appends -01 to generate the output file names.
#!/bin/bash
#SBATCH --qos=regular
#SBATCH --time=00:20:00
#SBATCH --job-name=prod
#SBATCH --reservation=amber26
#SBATCH --nodes=1
#SBATCH --gpus=1
#SBATCH --ntasks-per-node=1
#SBATCH --mem=10G
#SBATCH --output=prod-%j.out
#SBATCH --error=prod-%j.err
module load Amber/25.0-foss-2022a-AmberTools-25-CUDA-12.1.1-DeePMD
Base=${1}
Parm=${1}.parm7
mdin=mdins/11_Production_0.5ns.mdin
newname=${Base}-01
${AMBERHOME}/bin/pmemd.cuda_SPFP -O -p ${Parm} -c ${Base}.rst7 -i ${mdin} -o ${newname}.mdout -inf ${newname}.mdinfo -x ${newname}.nc -r ${newname}.rst7 -ref ${Base}.rst7
The core AMBER command is pmemd.cuda_SPFP, which runs GPU-accelerated
molecular dynamics. Its key flags are:
-O: overwrite existing output files-i: (input) control data for the MD run-o: (output) user readable state info and diagnostics-inf: (output) latest mdout-format energy info-p: (input) molecular topology, force field, periodic box type, atom and residue names-c: (input) initial coordinates and (optionally) velocities and periodic box size-ref: (input, optional) reference coords for position restraints-x: (output) coordinate sets saved over trajectory-r: (output) final coordinates, velocity, and box dimensions — used for restarting the run
2.2.5.3. Running the production simulation
Submit the production job:
sbatch prod.slurm MTR1_counterIon_water_bulk-run
The job should take approximately 10 minutes to complete. Output files
will be written with the prefix MTR1_counterIon_water_bulk-run-01.
Note
If you do not want to wait for the production job to finish, reference output files are available. Copy them to a subdirectory:
cp -r ../../output/Prod ./output
2.2.5.4. Wrapping and visualization of the production trajectory
Wrap the production trajectory:
cpptraj -i wrap_RNA_prod.in
Open the production trajectory in VMD locally. First, download the necessary files:
rsync -avz "username@hyperion.sw.ehu.es:/scratch/amber26-XX/TutorialData/HandsOn2_MD/working_dir/Prod/{centered.MTR1_counterIon_water_bulk-run-01.nc,MTR1_counterIon_water_bulk-run.parm7,MTR_waterreps.txt}" .
Locally: Open the trajectory in VMD:
vmd -e MTR_waterreps.txt \
centered.MTR1_counterIon_water_bulk-run-01.nc \
MTR1_counterIon_water_bulk-run.parm7
Inspect the dynamics of the different components. Below is a movie demonstration:
Warning
This was a very short production simulation. Realistically, it would be desirable to perform multiple independent long production simulations. In previous studies of MTR1, five independent production simulations of 150 ns each were performed. Analysis of the root mean square deviation (RMSD) indicated that the first 50 ns corresponded to equilibration, while the remaining 100 ns of each simulation were used for production analysis, resulting in a total of 500 ns of production data.
The figure below shows the RMSD of the RNA heavy atoms with respect to the first trajectory frame over four independent 150 ns production simulations.
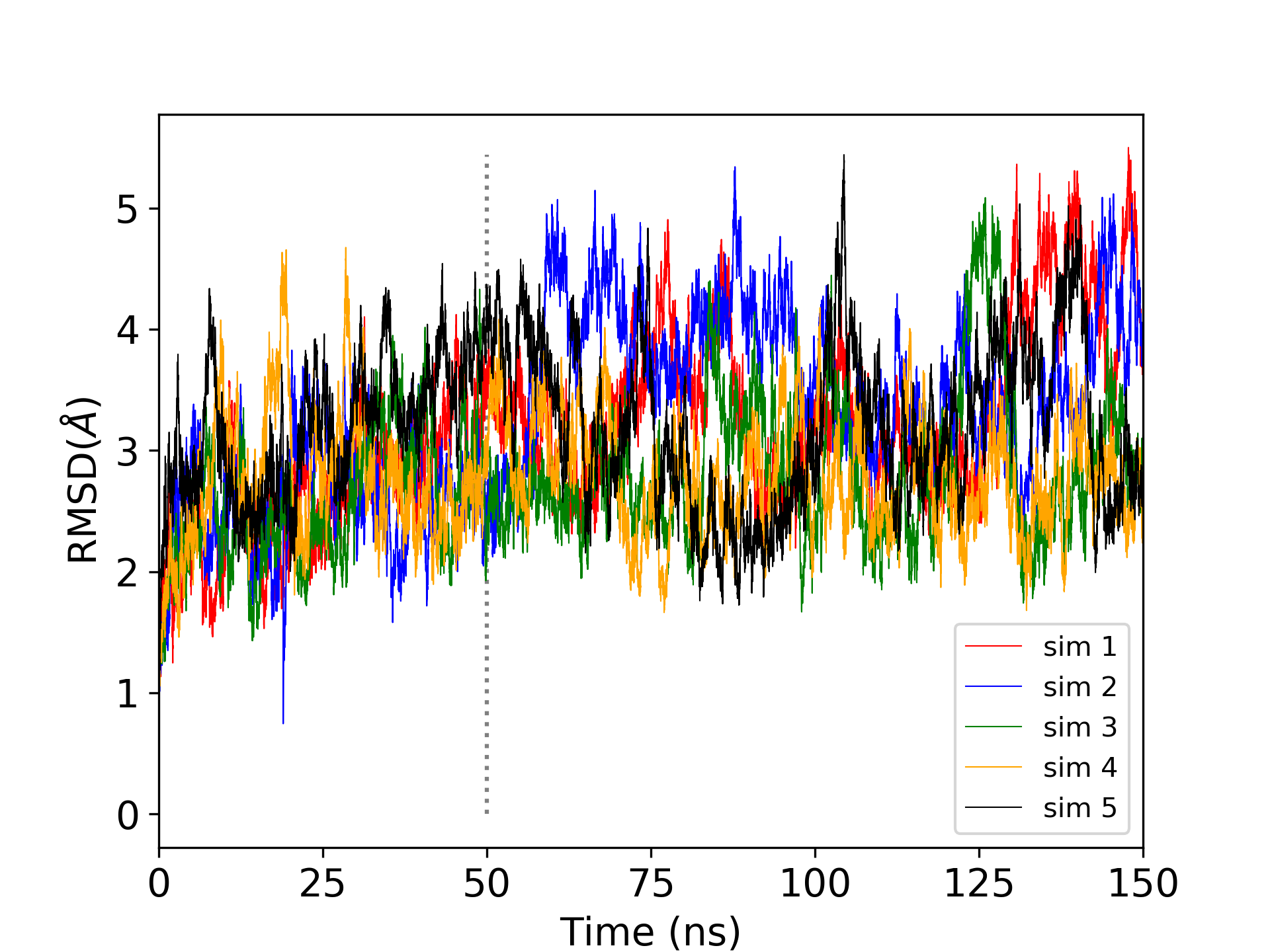
Figure 3. Root mean square deviation of the positions of heavy atoms with respect to the first trajectory frame over five independent 150 ns production simulations.
Hint
You can compute the RMSD of your own trajectory using the following
cpptraj input file. The rms command calculates
the RMSD of all RNA heavy atoms (hydrogens excluded, !@H=) with
respect to the first frame (first). The mass keyword enables
mass-weighting:
parm MTR1_counterIon_water_bulk-run.parm7
trajin centered.MTR1_counterIon_water_bulk-run-01.nc
rms ToFirst :1-69&!@H= first out rmsd1.dat mass
run
quit
cpptraj -i rmsd.in
2.2.6. (Optional) Additional Exercise: Performing a brief energy minimization
For this optional exercise, change directories back to the equilibration directory:
cd ../Equil
The following exercise should only be performed if you have time left over
or are waiting for other simulations to run. In this exercise, you will run the 000_Min step of the equilibration
procedure independently using 000_Min.mdin.
You have already been introduced to the equilibration script
eqFromStratch.slurm. In this optional exercise, you will modify this
script to run only the initial minimization step on the CPU.
Copy the equilibration script and create a new script for the minimization only run:
cp eqFromStratch.slurm eqFromStratch_000.slurm
Open eqFromStratch_000.slurm in a text editor (e.g. vim) and modify
the SLURM header so that the job runs on the CPU rather than on a GPU:
#!/bin/bash
#SBATCH --qos=regular
#SBATCH --time=00:30:00
#SBATCH --job-name=min
#SBATCH --reservation=amber26
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --mem=10G
#SBATCH --output=equil-%j.out
#SBATCH --error=equil-%j.err
Next, edit the script so that only the 000_Min step is executed. Delete
all other steps and retain only the following definition:
steps="\
000_Min \
"
In this case, only sander will be invoked, so no GPU resources are
required. The job is expected to finish in approximately 10 minutes.
The Equil directory should then contain the following:
ls
MTR1_counterIon_water_bulk.parm7
MTR1_counterIon_water_bulk.rst7
eqFromStratch.slurm
eqFromStratch_000.slurm
mdins
restrnt.disang
wrap_RNA_equil.in
Submit the job:
sbatch eqFromStratch_000.slurm MTR1_counterIon_water_bulk
When the job is complete, monitor how the total energy (EAMBER)
decreases over the course of the minimization:
grep -r EAMBER 000_Min-run.mdout | awk '{print $3}' | head -n10 > ene.dat
Plot the energy using xmgrace. Load the module first:
module load Grace
xmgrace ene.dat
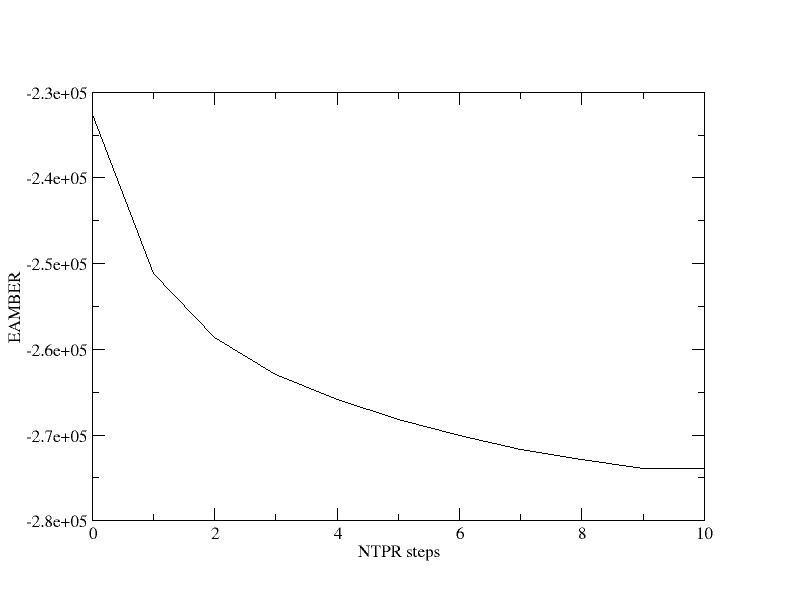
Figure 4. Total energy output every 50 optimization steps during
the 000_Min minimization.
You will observe that the total energy (in kcal/mol) decreases rapidly at the beginning of the minimization and then begins to plateau. In this run, the energy was printed every 50 minimization steps.
Open 000_Min-run.mdout in a text editor and inspect the individual energy components:
NSTEP ENERGY RMS GMAX NAME NUMBER
1 1.7197E+06 6.7827E+04 2.2501E+07 N3 1730
BOND = 5038.2053 ANGLE = 397.2193 DIHED = 1511.0059
VDWAALS = 1926187.1298 EEL = -206390.2786 HBOND = 0.0000
1-4 VDW = 697.4115 1-4 EEL = -7778.5268 RESTRAINT = 0.0000
NSTEP ENERGY RMS GMAX NAME NUMBER
50 -2.3201E+05 4.6787E+00 6.6797E+02 C6 1723
BOND = 22954.6607 ANGLE = 746.4316 DIHED = 1649.0907
VDWAALS = 27936.2925 EEL = -278521.9124 HBOND = 0.0000
1-4 VDW = 650.9327 1-4 EEL = -7954.8135 RESTRAINT = 533.4714
EAMBER = -232539.3177
You will notice that the van der Waals energy decreases rapidly at the beginning of the minimization as bad contacts are relieved. This illustrates the importance of a gradual and well-controlled equilibration procedure.
2.3. Relevant literature
References cited in this tutorial are listed below.