8. Hands-On Session 8: Analyzing Alchemical Free Energy Results Using FE-Toolkit
8.1. Learning Objectives
8.2. Activities
flowchart LR
%% ===== extract simulation outputs to dats =====
A1["Amber AFE outputs<br/>mdouts<br/>rem.log<br/>rest.in"]
P1{{"<b>[§8.2.1.1]</b><br/>Extract per-lambda data<br/>edgembar-amber2dats.py"}}
O1["<b>[from §8.2.1.1]</b><br/>efep dat files<br/>edge/env/stage/trial dats"]
A1 --> P1
P1 --> O1
%% ===== build edge xml and run edgembar =====
P2{{"<b>[§8.2.1.2]</b><br/>Discover edges, write xml<br/>DiscoverEdges.py"}}
O2["<b>[from §8.2.1.2]</b><br/>Edge input<br/>edge_ejm31.xml"]
P3{{"<b>[§8.2.1.2]</b><br/>Run BAR/MBAR analysis<br/>edgembar_omp"}}
O3["<b>[from §8.2.1.2]</b><br/>Report script and data<br/>edge_ejm31.py<br/>edge_ejm31.html<br/>edge_ejm31.nc"]
O1 --> P2
P2 --> O2
O2 --> P3
P3 --> O3
%% ===== interpret html report =====
P4{{"<b>[§8.2.2]</b><br/>Interpret edge report<br/>web browser"}}
O4["<b>[from §8.2.2]</b><br/>Convergence assessment<br/>BAR/TI dG estimates"]
O3 --> P4
P4 --> O4
%% ===== read nc with python api =====
P5{{"<b>[§8.2.3]</b><br/>Parse edge data<br/>edgembar Python API"}}
O5["<b>[from §8.2.3]</b><br/>dG / ddG estimates<br/>portable analysis"]
O3 --> P5
P5 --> O5
classDef file fill:#fff7e6,stroke:#d98c00,stroke-width:1.5px,color:#111;
classDef program fill:#e8f1ff,stroke:#1f77b4,stroke-width:1.8px,color:#111;
classDef result fill:#eaf7ea,stroke:#2ca02c,stroke-width:1.5px,color:#111;
class A1 file;
class P1,P2,P3,P4,P5 program;
class O1,O2,O3,O4,O5 result;
In HandsOn9 and HandsOn10, you will learn how to run different variations of alchemical free energy simulations. In this HandsOn tutorial, you will learn how to analyze the results of these simulations using FE-Toolkit.
You will learn how to extract data from simulation output files, generate analysis reports for relative and absolute binding free energy calculations, and interpret the results.
8.2.1. Getting Started with FE-Toolkit
For this tutorial, we have provided the simulation output data from Absolute Binding Free Energy Calculations on Tyk2 for four ligands. In this tutorial, we will primarily use the files for the ligand ejm31; however, we have provided the other ligands as a reference (and as an opportunity for further practice).
To obtain the files, download this ABFE Simulation Output. This file is quite large - so it may take a few minutes to download.
Hint
The FE-Toolkit paper published by Giese and co-workers includes a significant Supporting Information that outlines much of what is included in this tutorial, as well as the underlying math driving these calculations. See the paper Giese et al.[1] for more information.
8.2.1.1. Simulation Outputs from Alchemical Free Energy Simulations with Amber
The fundamental outputs from an Absolute Binding Free Energy (ABFE) calculation that we will be using are the following:
mdouts: These are Amber simulation output files that include energy information from each lambda window.
rem.log: These are Amber simulation output files that include information about replica exchange acceptances.
rest.in (ABFE Only): This file contains information about the Boresch restraints used in the Amber simulation.
Briefly - we will explore each of these files:
Amber AFE calculations write MDouts with entries that look something like this. They include information about the energies, the dV/dL value, which exchange you are on, as well as information about the soft-core potentials.
| TI region 1
NSTEP = 840250 TIME(PS) = 3361.000 TEMP(K) = 294.22 PRESS = 0.0
Etot = -33055.3946 EKtot = 6151.6533 EPtot = -39207.0479
BOND = 0.0000 ANGLE = 0.0000 DIHED = 3.6590
1-4 NB = -0.0696 1-4 EEL = -25.0360 VDWAALS = 6648.6562
EELEC = -45834.2575 EHBOND = 0.0000 RESTRAINT = 0.0000
DV/DL = 79.6779
TEMP0 = 298.0000 REPNUM = 9 EXCHANGE# = 8403
------------------------------------------------------------------------------
Softcore part of the system: 32 atoms, TEMP(K) = 269.57
SC_Etot= 41.8564 SC_EKtot= 22.7663 SC_EPtot = 19.0901
SC_BOND= 6.4851 SC_ANGLE= 11.9679 SC_DIHED = 6.5738
SC_14NB= 0.0000 SC_14EEL= 0.0000 SC_VDW = -5.9367
SC_EEL = 0.0000
SC_RES_DIST= 0.0000 SC_RES_ANG= 0.0000 SC_RES_TORS= 0.0000
SC_EEL_DER= 1.1039 SC_VDW_DER= -15.3796 SC_DERIV = -14.2757
------------------------------------------------------------------------------
| TI region 2
NSTEP = 840250 TIME(PS) = 3361.000 TEMP(K) = 294.32 PRESS = 0.0
Etot = -33078.1609 EKtot = 6128.8870 EPtot = -39207.0479
BOND = 0.0000 ANGLE = 0.0000 DIHED = 3.6590
1-4 NB = -0.0696 1-4 EEL = -25.0360 VDWAALS = 6648.6562
EELEC = -45834.2575 EHBOND = 0.0000 RESTRAINT = 0.0000
DV/DL = 79.6779
TEMP0 = 298.0000 REPNUM = 9 EXCHANGE# = 8403
------------------------------------------------------------------------------
Amber also writes rem.log files which list the acceptances in exchanges between windows.
# Replica Exchange log file
# numexchg is 12500
# REMD filenames:
# remlog= remd_complex_ejm31.log
# remtype= rem.type
# Rep#, Neibr#, Temp0, PotE(x_1), PotE(x_2), left_fe, right_fe, Success, Success rate (i,i+1)
...
# exchange 9032
1 2 298.00 -39400.10 -39409.58 -Infinity -1.79 T 0.87
2 1 298.00 -39408.05 -39398.45 1.79 -10.40 T 0.35
3 4 298.00 -39416.77 -39352.95 10.40 -22.46 F 0.06
4 3 298.00 -39332.22 -39390.84 22.45 -33.45 F 0.01
5 6 298.00 -39316.56 -39355.34 32.76 -36.65 F 0.00
6 5 298.00 -39321.48 -39275.80 36.74 -34.91 F 0.00
7 8 298.00 -39129.67 -39121.37 32.64 -25.37 F 0.00
8 7 298.00 -39108.33 -39103.97 23.32 -13.02 F 0.00
9 10 298.00 -39087.24 -39262.19 10.73 -3.20 F 0.07
10 9 298.00 -39262.99 -39081.81 3.20 -0.44 F 0.74
11 -1 298.00 -39197.79 0.00 0.03 0.00 F 0.00
# exchange 9033
1 -1 298.00 -39438.52 0.00 -Infinity -1.79 F 0.87
2 3 298.00 -39467.30 -39324.55 1.79 -10.40 T 0.35
3 2 298.00 -39314.20 -39456.10 10.40 -22.46 T 0.06
4 5 298.00 -39259.84 -39308.14 22.45 -33.45 F 0.01
5 4 298.00 -39282.02 -39223.52 32.76 -36.65 F 0.00
6 7 298.00 -39248.23 -39241.31 36.74 -34.91 F 0.00
7 6 298.00 -39220.57 -39210.90 32.64 -25.37 F 0.00
8 9 298.00 -39378.26 -39145.48 23.32 -13.02 F 0.00
9 8 298.00 -39137.02 -39358.97 10.73 -3.20 F 0.07
10 11 298.00 -39218.70 -39185.63 3.20 -0.44 T 0.74
11 10 298.00 -39185.17 -39218.08 0.03 0.00 T 0.00
Lastly, in ABFE we use rest.in files to define Boresch restraints (see the Boresch Restraint Tutorial) which hold the ligand in the protein while its interactions are decoupled from the rest of the system at \(\lambda=1\). These restraint files contain the 0-indexed atoms for a pairwise restraint, two angle restraints, and three dihedral restraints between the protein and the ligand.
&rst iat=1489,15
r1=0.00000,r2=4.44575,r3=4.44575,r4=999.000,rk2=15.06, rk3=15.06/
&rst iat=1477,1489,15
r1=-180.00000,r2=83.99734,r3=83.99734,r4=180.000,rk2=53.21, rk3=53.21/
&rst iat=1489,15,14
r1=-180.00000,r2=66.38281,r3=66.38281,r4=180.000,rk2=40.55, rk3=40.55/
&rst iat=1490,1477,1489,15
r1=-180.00000,r2=11.03062,r3=11.03062,r4=180.000,rk2=29.83, rk3=29.83/
&rst iat=1477,1489,15,14
r1=-180.00000,r2=30.50819,r3=30.50819,r4=180.000,rk2=62.83, rk3=62.83/
&rst iat=1489,15,14,16
r1=-180.00000,r2=-137.04995,r3=-137.04995,r4=180.000,rk2=49.20, rk3=49.20/
Note
Boresch restraints do work on the system during an ABFE calculation, and thus have to be included when analyzing them. These will be handled automatically by edgembar during this tutorial, but you can learn more on their impact on the free energy from (see the Boresch Contributions to the Free Energy Tutorial).
Take a look at the folders you downloaded above, and take a minute to look through these files. For each ligand, you have a rem.log file, many mdouts (1/\(\lambda\)-window), and one rest.in file. You would also normally have a trajectory output for at least the end-states; however, that is out of the scope for the present tutorial.
8.2.1.2. Generating Edgembar HTML Reports
For this tutorial, you need to have edgembar installed. This can either be installed from source (https://gitlab.com/RutgersLBSR/fe-toolkit); however, there is also a Python distribution of edgembar (the component of FE-Toolkit we will be using) available on PyPI (pip install edgembar). More edgembar documentation is located here: https://rutgerslbsr.gitlab.io/fe-toolkit/edgembar/index.html
By default, the Edgembar program reads files that are program agnostic (dat files of the format column1: simulation time, column2: potential energy in kcal/mol). It generally expects these files to be split into the following substructure:
edge/
env/
stage/
trial/
Note
edge: Represents the overall ABFE/RBFE calculation. The name comes from thinking of the calculation as an edge in an alchemical network.
env: This is the environment (e.g. aqueous, complex)
stage: This is what stage for that environment (for instance, if you have separate charge decoupling and vdw decoupling stages). With SmoothStep potentials, often there is just a single vdw stage.
trial: A subdirectory for each independent trial, usually named t1 t2 etc.
In a way, these are just labels. The important thing is that there is a single edge, two env values, and there can be arbitrarily many stage and trial values. Each of them represents a calculation, and a very raw sketch of their relationship would look like this:
In the case of using Amber as the simulation package, FE-Toolkit has built-in tools for generating this hierarchical structure, specifically edgembar-amber2dats.
For this - we will use a short bash script.
edge=$1
mkdir -p dats
for env in {complex,binder};
do
for stage in vdw;
do
for trial in {t1,t2};
do
echo "Extracting files for $edge/$env/$stage/$trial"
mkdir -p dats/$edge/$env/$stage/$trial
if [ "$env" == "complex" ];
then
edgembar-amber2dats.py -r $edge/$trial\_$env/remd_$edge.log --odir dats/$edge/$env/$stage/$trial/ --vba $edge/$trial\_$env/rest.in $(ls $edge/$trial\_$env/*mdout) &
else
edgembar-amber2dats.py -r $edge/$trial\_$env/remd_$edge.log --odir dats/$edge/$env/$stage/$trial/ $(ls $edge/$trial\_$env/*mdout) &
fi
done
done
done
Let’s explore the edgembar-amber2dats.py script quickly before running the script.
edgembar-amber2dats.py -h
There are a few important options that you should consider:
nan: What should MBAR do if an MBAR energy is ‘**’. The default is to revert to BAR analysis. Important for ABFE.
extra: Can add extra data to the edgembar output for decomposing DV/DL profiles. Its main use is plotting the contributions from electrostatics, 1-4 vdw interactions, etc.
For now, we will run without these.
Run the script for ejm31.
bash run_amber2dats.sh ejm31
Now - you’ll see lots of messages saying something along the lines of “Invalid energies within ejm31/t2_complex/complex_ejm31_1.00000.mdout; writing output for BAR. See the –nan option”. This is expected for ABFE, because for MBAR you evaluate the energy at every lambda point with respect to every other lambda point. For ABFE, this means that you often can be evaluating energies of clashes between the protein and the ligand (for instance, in a state where your ligand is fully decoupled.)
Now that these files are generated, take a moment to explore the dats folder. Note that it follows the rough scheme we described above of edge/env/stage/trial. Take a look at the files that you have there.
Now, we will use a small python script to create an xml for edgembar to work on.
#!/usr/bin/env python3
import edgembar
import os
from pathlib import Path
odir = Path("analysis")
s=r"dats/{edge}/{env}/{stage}/{trial}/efep_{traj}_{ene}.dat"
exclusions=None
# Load the dat files into an edges object
edges = edgembar.DiscoverEdges(s,
exclude_trials=exclusions,
target="complex",
reference="binder")
odir.mkdir(exist_ok=True)
for edge in edges:
# Tell the edges about the shift from the boresch restraints.
edge.SetVBAShift()
# Reverse the order of lambdas (1->0) so that the sign is negative, which is what you would expect for an ABFE.
for trial in edge.GetAllTrials():
trial.reverse()
fname = odir / (edge.name + ".xml")
edge.WriteXml(fname)
A few things are happening in this file. The first is we are setting a template string to say how the dats folder is structured. Then we generate an edges object with DiscoverEdges. If you have outliers, you could potentially want to exclude those trials using the exclusions list (e.g. exclusions=[“t1”])
Now - run this script.
python DiscoverEdges.py
You’ll see a new analysis directory with xml files present for each of the transformations you ran amber2dats for previously (up to four!). These .xml files are the input to edgembar. Open one of them and you’ll see they have the same hierarchical structure of edges, environments, stages, and trials, along with the lambda values per trial (yes, you can use different schedules in every trial of every stage and environment), the path to the .dat files, and an optional constant shift in the free energy, if you used boresch restraints for that trial.
Next, run the actual BAR/MBAR calculation:
OMP_NUM_THREADS=8 edgembar_omp analysis/edge_ejm31.xml --fwdrev --halves
Warning
edgembar and edgembar_omp are dependent on relative paths defined in the xml file. Thus, you must run it from outside of analysis.
The two options enable additional analysis for edgembar that we will discuss in a later part of this tutorial when we are looking at the HTML reports.
This script writes a Python script, which we can run as:
python analysis/edge_ejm31.py
python analysis/edge_ejm31.py --netcdf
Note that we are running it twice. If you only want the HTML report, the first command is enough; however, if you want a portable data file you can pass to other programs or parse yourself, the NetCDF option provides a self-contained file for you to do so.
Now you should see edge_ejm31.html and edge_ejm31.nc in your analysis directory!
8.2.2. FE-Toolkit HTML Reports
For this tutorial, we have provided the simulation output data from Absolute Binding Free Energy Calculations on Tyk2 for four ligands. In this tutorial, we will primarily use the files for the ligand ejm31; however, we have provided the other ligands as a reference (and as an opportunity for further practice).
The HTML report can be viewed here: Graph.html
8.2.2.1. Header
Hint
** Troubleshooting Questions **
Do you have many errors and warnings?
Are you using a recent hash of edgembar?
The first thing you see in an HTML report is the header. It contains important information about the calculation, specifically which version of edgembar was used, what command called edgembar, and what errors and warnings were generated.

A few things are worth pointing out: every version of edgembar is given a unique hash (in the above image 3.6.6 H09ccf5), which can be useful for debugging.
Next, a summary of errors is included. These are hyperlinks to the data where the errors occurred. Usually these correspond to less-than-ideal equilibration, although they are not necessarily fully impactful. Errors are shown in red and warnings are shown in yellow.
8.2.2.2. Objective Function
The next plot you see in the edge report is the Objective Function that was minimized across all samples to obtain the final value. This plot is probably the least informative, until you run into a case where your objective function does not look like a parabola. That’s a sign that things went very wrong with the simulation.
8.2.2.3. Overall results
Hint
** Troubleshooting Questions **
Do your BAR and TI results in the table agree?
How far does your fwdrev analysis deviate from production?
Is there a significant trend in your first and last half analysis?
The next set of data you’ll see is a table that includes the values of the free energy (and if you ran edgembar with –fwdrev and –halves, some other analysis too!).
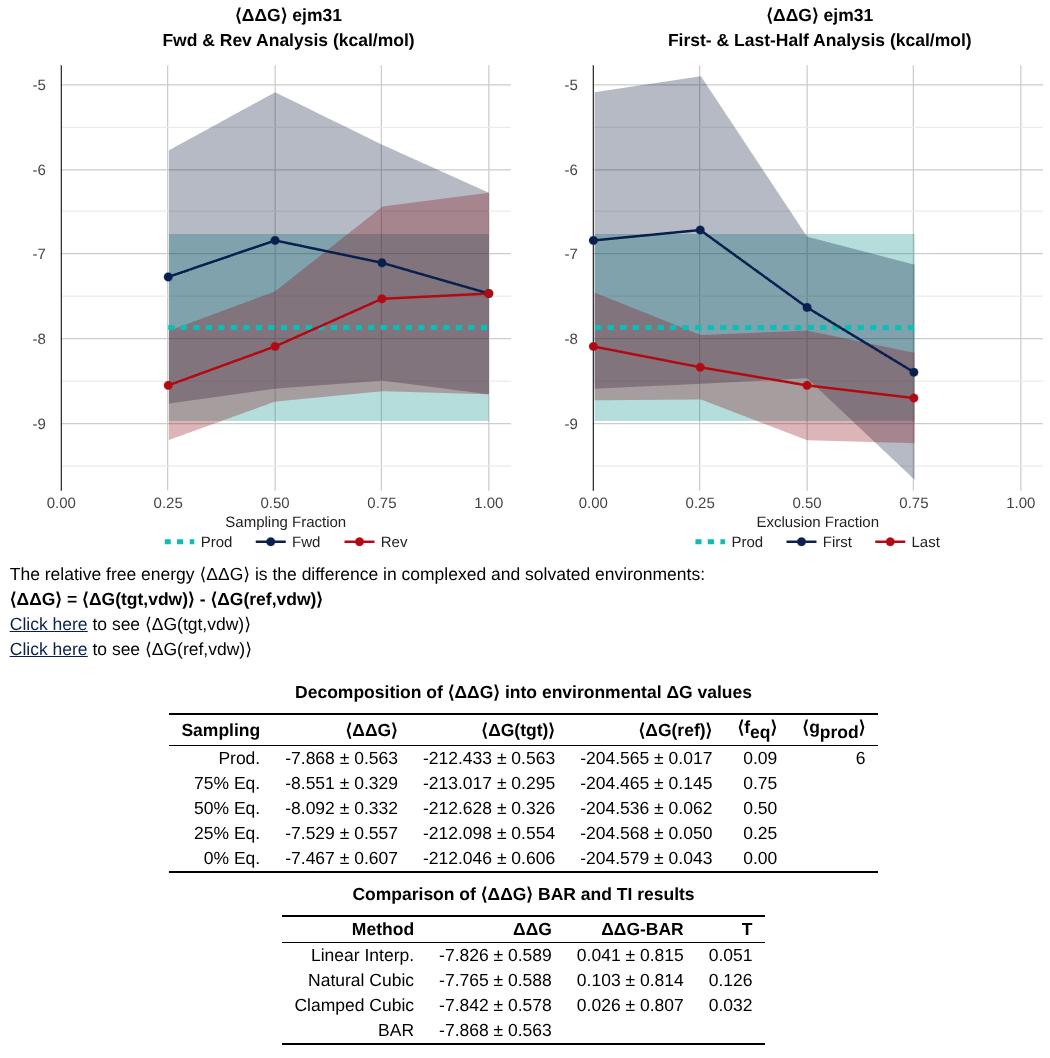
Most important is the table at the bottom, which gives you a summary of the values from analysis with Bennett’s Acceptance Ratio (BAR), and Thermodynamic Integration (TI).
Note
A key debugging tool is to check agreement between BAR and TI. If these values do not agree - it suggests a problem with either equilibration, replica exchange acceptances, or alchemical pathway.
With –fwdrev and –halves, you get the two plots at the top of the image above which include timeseries analysis.
The fwdrev analysis compares the free energies calculated from the first X% of the data to those calculated from the last X% of the data.
The halves analysis excludes X% of the simulation from start as equilibration, and splits the remaining 100-X% of data into two halves and compares the free energy from each half.
8.2.2.4. DV/DL Profiles and Replica Exchanges
Hint
** Troubleshooting Questions **
Is your DV/DL profile smooth? Does it have weird discontinuities?
Are there big differences in your DV/DL profiles between trials?
Does your replica exchange acceptance ratio ever approach a low number (< 0.2?)
The next key set of plots that are found are the DV/DL profile, and the Replica Exchange Acceptance Ratios.
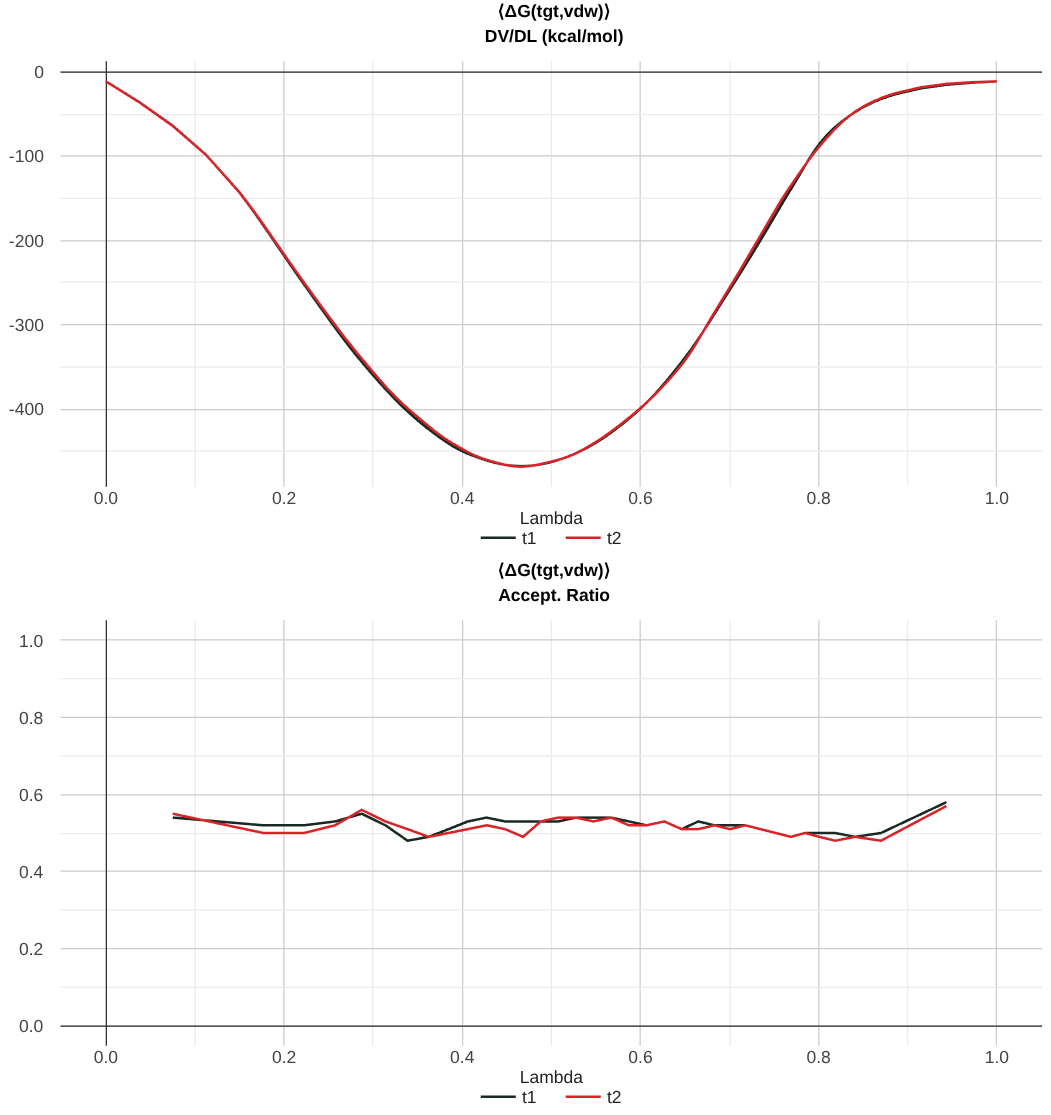
In the above image, you can see a well-converged DV/DL profile. Note that the trials track pretty well along each other.
Recall that the dV/dL profile is intrinsically linked to the free energy by the equation:
Note
For ABFE calculations, the dV/dL profile is shifted by a constant number. This number originates from the correction to the free energy that comes from Boresch Restraints. Determining the Boresch Restraint Contribution to the Free Energy.
Now, the replica exchange acceptance ratio is included in the second plot, and it gives you information for each pair of lambda windows what percentage of exchanges were accepted. Here, the value of 0.5 suggests that there were sufficient exchanges.
8.2.2.5. Data Tables
Hint
** Troubleshooting Questions **
Did a lot of simulation data get thrown out when selecting production regions?
Did this trial have many (or most) of the warnings?
Do you have single passes and round trips in your calculation?
The next major section of the file is a set of tables (per environment and per trial) that give summary information about the free energy calculation as a function of lambda.
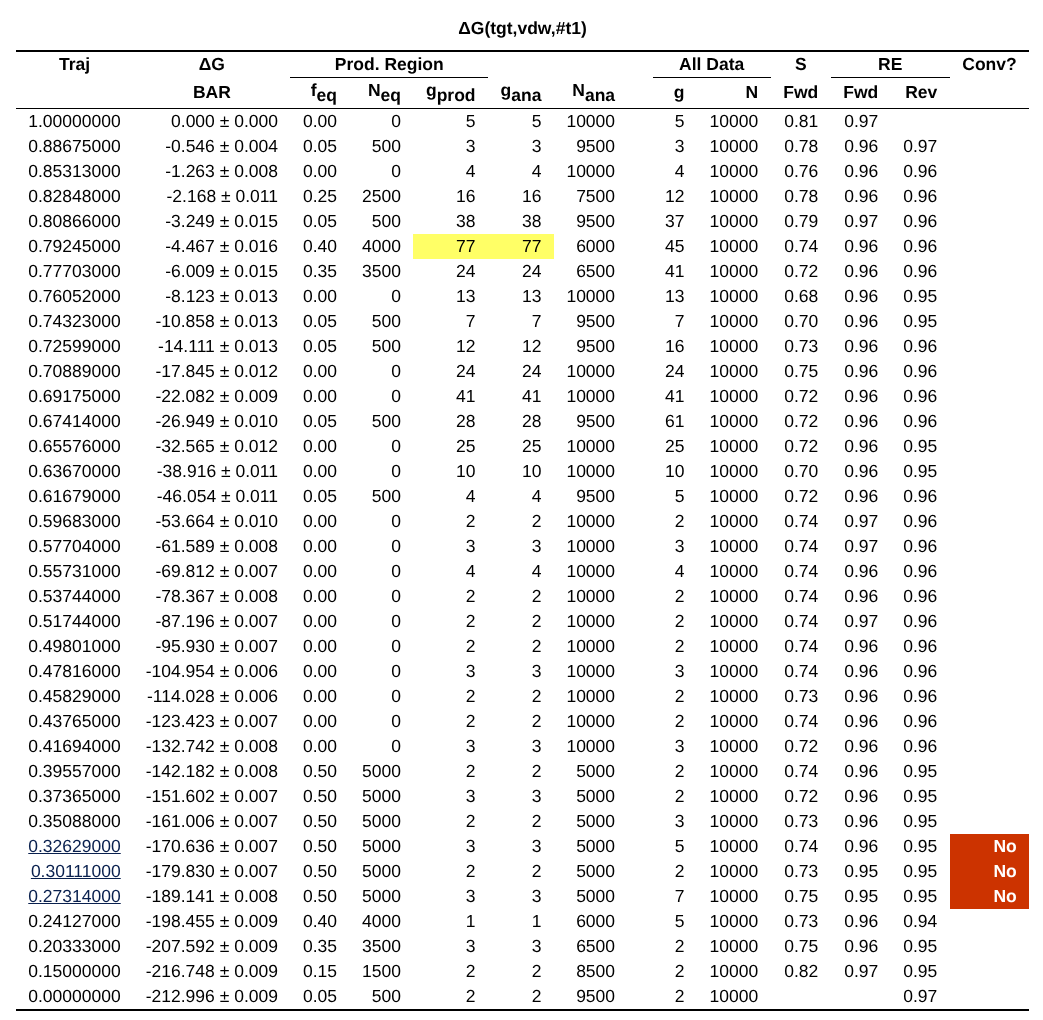
There is also a replica exchange data table that provides the data included in the acceptance ratio plot.
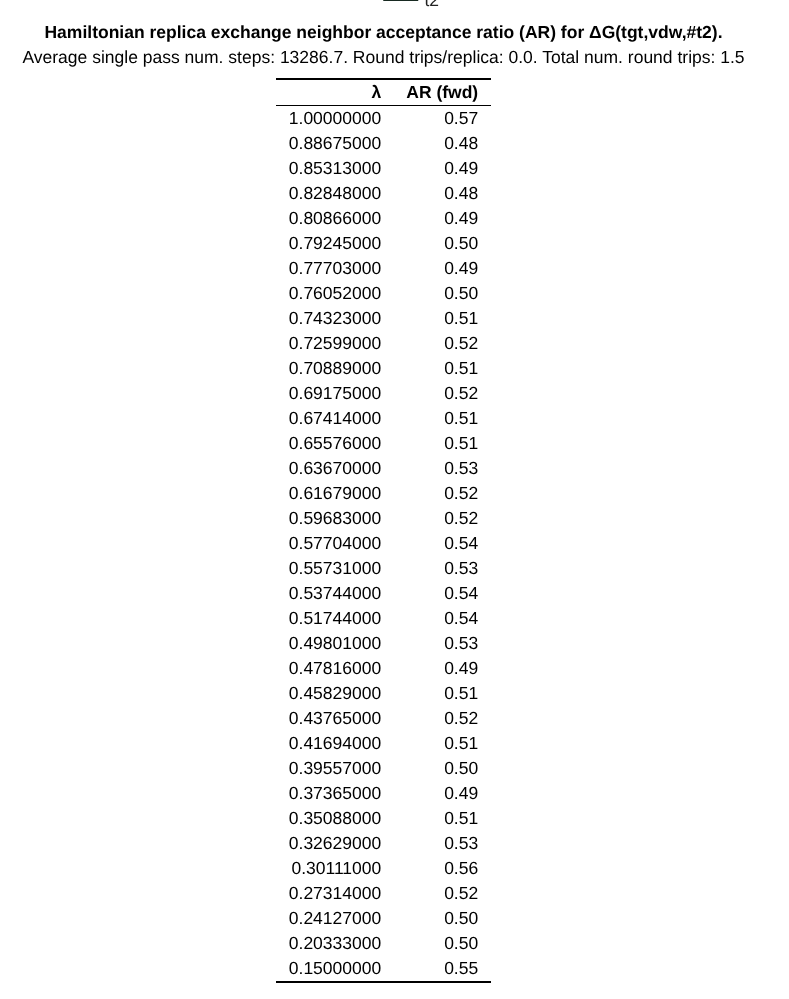
The header of this table includes a few additional useful pieces of information:
Average single pass num. steps: The amount of time it takes for a replica to exchange all the way \(0 \rightarrow 1\) or \(1 \rightarrow 0\)
Round trips/replica: Total number of round trips divided by the number of replicas.
Total num. round trips: The number of times a replica traverses \(0 \rightarrow 1 \rightarrow 0\) or \(1 \rightarrow 0 \rightarrow 1\)
8.2.2.6. Troubleshooting
Hint
** Troubleshooting Questions **
Does the window look converged?
What is the scale of the dependence on the y-axis of the block average
Each lambda window that has a warning also gets a special section which includes information about the convergence of that window.
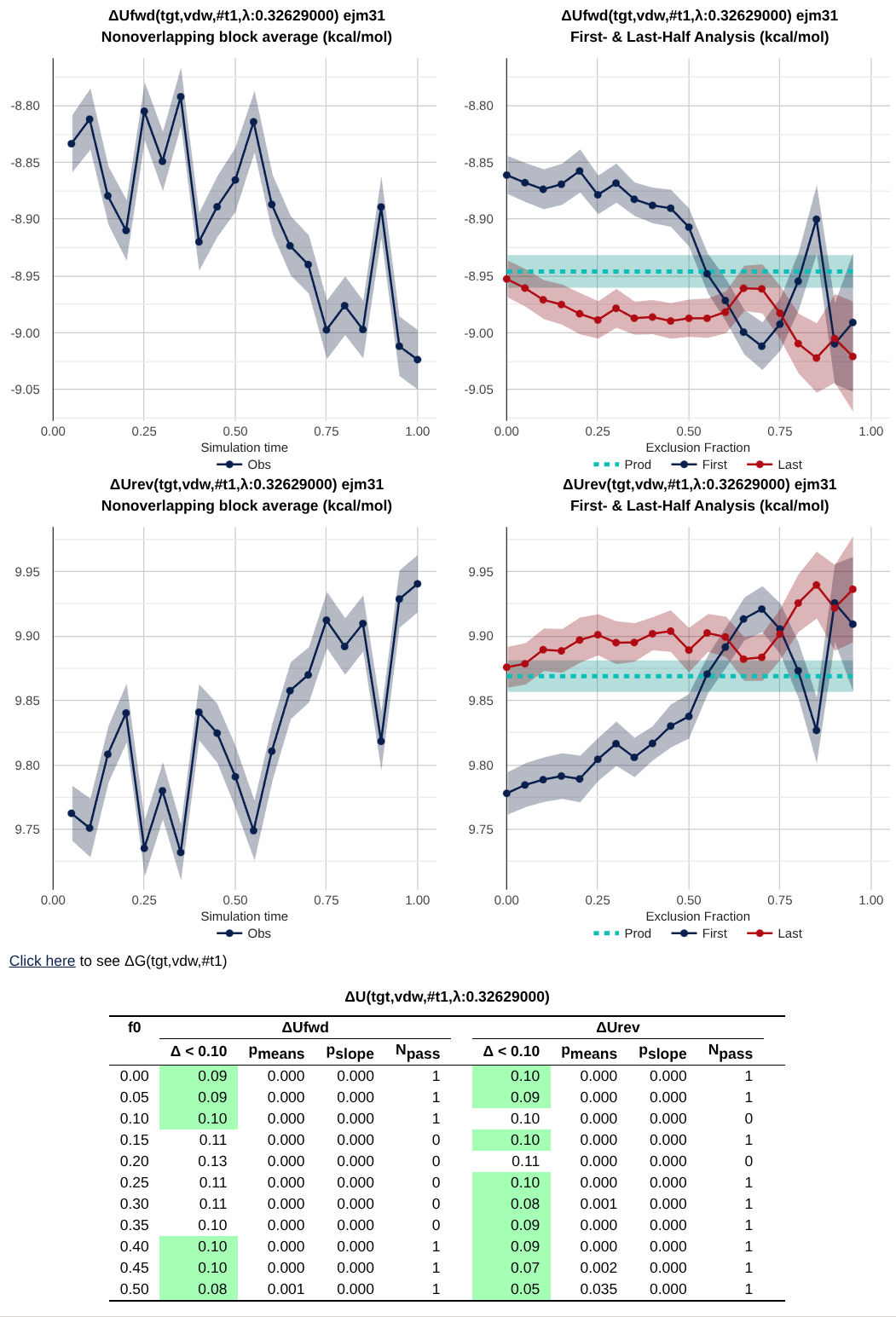
This shows plots of block averages over time, data tables, etc that can be used for additional debugging.
8.2.2.7. Wrapping Up
In this activity, you walked through the pieces of an HTML report. Go through the troubleshooting questions at the top of each relevant section with that report. Does this pass as an okay simulation?
8.2.3. Introduction to the Edgembar Python API
Data generated from the edgembar package in FE-Toolkit Giese et al.[1] is typically stored in one of three sources. The first is an edge Python file (e.g. edge_ejm31.py), the second is a NetCDF file (e.g. edge_ejm31.nc), and the third is an HTML report (e.g. edge_ejm31.html). Each serves a particular purpose; however, when developing outside analysis tools that interact with them, the choice can make a significant impact. For instance, the .py file is dependent on relative paths, so if you move it to a different directory, then it will no longer be able to generate an HTML report. The HTML report includes a significant amount of information and is portable; however, it’s challenging to parse in Python. Therefore, the NC file exists as a portable (and parseable) solution.
To start, download the NetCDF file for an ABFE run on ejm31: This file
from __future__ import annotations
import argparse
from pathlib import Path
from edgembar.HtmlUtils import GetReplExchData
from edgembar.NcIO import LoadEdgeNc
def format_energy(value: float, error: float) -> str:
return f"{value:8.3f} +- {error:6.3f} kcal/mol"
def print_bar_summary(label: str, obj, prod_data) -> None:
value, error = obj.GetValueAndError(prod_data)
print(f"{label:<28} {format_energy(value, error)}")
def print_ti_summary(edge) -> None:
ti_data = edge.GetTIValuesAndErrors()
if ti_data is None:
print("TI estimates: unavailable")
return
print("TI estimates:")
for method in ("Linear", "Natural", "Clamped"):
if method in ti_data:
value, error = ti_data[method]
print(f" {method:<8} {format_energy(value, error)}")
def print_message_summary(edge) -> None:
messages = edge.GetErrorMsgs()
errors = [msg for msg in messages if msg.iserr]
warnings = [msg for msg in messages if not msg.iserr and msg.kind != "outlier"]
print(f"Errors: {len(errors)}")
print(f"Warnings: {len(warnings)}")
def print_trial_summary(trial, prod_data) -> None:
print_bar_summary(f" trial {trial.name}", trial, prod_data)
rem_data = GetReplExchData(trial)
if rem_data is None:
return
single_pass = rem_data["Average single pass steps:"]
trips_per_replica = rem_data["Round trips per replica:"]
total_round_trips = rem_data["Total round trips:"]
print(
" " * 8
+ "RE summary: "
+ f"single-pass={single_pass:.1f}, "
+ f"round-trips/replica={trips_per_replica:.2f}, "
+ f"total-round-trips={total_round_trips:.1f}"
)
def build_parser() -> argparse.ArgumentParser:
parser = argparse.ArgumentParser(
description="Read an EdgeMBAR .nc file and print a simple energy summary."
)
parser.add_argument("edge_nc", type=Path, help="Path to an EdgeMBAR NetCDF file")
return parser
def main() -> None:
args = build_parser().parse_args()
if args.edge_nc.suffix != ".nc":
raise SystemExit("Expected a NetCDF edge file ending in .nc")
if not args.edge_nc.exists():
raise SystemExit(f"File not found: {args.edge_nc}")
edge = LoadEdgeNc(str(args.edge_nc))
prod_data = edge.results.prod
print(f"Edge: {edge.name}")
print_bar_summary("edge", edge, prod_data)
print_ti_summary(edge)
print_message_summary(edge)
print()
for env in edge.GetEnvs():
print_bar_summary(f" env {env.name}", env, prod_data)
for stage in env.stages:
print_bar_summary(f" stage {stage.name}", stage, prod_data)
for trial in stage.trials:
print_trial_summary(trial, prod_data)
print()
if __name__ == "__main__":
main()
Above is a Python script that you can use to access the data that you were looking at in the HTML report from the NetCDF file.
The key steps are:
Load an edge with
LoadEdgeNcThe free energy is obtained by running
edge.GetValueAndError(edge.results.prod)(orstage, orenv, ortrial)The replica exchange information is provided by
GetReplExchData(trial)