3. Hands-On Session 3: Build an RNA enzyme (ribozyme) system containing 12-6-4 divalent ions and non-standard residues
3.1. Learning Objectives
At the end of this hands-on session, you will be able to:
Generate AMBER topology and coordinate files for molecular dynamics simulations
Identify and resolve common atom-type errors encountered during system building
Incorporate non-standard nucleic acid residues using modXNA
Use the RESP procedure to parameterize a non-standard residue
Determine MM charges from an electrostatic potential calculation
Build a solvated RNA system in LEaP under physiologically relevant conditions
Explain why an accurate description of ion-nucleic acid interactions is important
Introduce divalent ions described by the 12-6-4 Lennard-Jones potential
Apply 12-6-4 parameters and the PGY pairwise corrections in Amber
3.2. Activities
Note
This activity is designed to teach you how to build a system using AMBER and is based on the MTR1 ribozyme that you examined yesterday. However, for pedagogical reasons, the system has been prepared differently here.
The simulations presented yesterday refer to papers published in 2022 and 2023 [1][2], at which time the modXNA parameters (released in 2024) for the parametrization of non-standard residues were not yet available. Therefore, non-standard residues were parametrized using the classical Antechamber and RESP procedure [3].
In this activity, we will focus primarily on the more recent modXNA parametrization approach. The classical Antechamber/RESP procedure will be treated as an additional exercise, since it will be covered in detail in another activity at a later stage, particularly in the context of free energy calculations and ligand parametrization.
In addition, for pedagogical purposes, we demonstrate how to apply the Panteva 12-6-4 correction terms for magnesium–nucleic acid interactions. Since no magnesium ions were initially present in the MTR1 structure, the original barium ions were replaced with magnesium ions.
3.2.1. Accessing the Tutorial Files
To get the tutorial data during the workshop, you can get a copy into your scratch with this command.
DataDir="/data/amber26/TutorialData/HandsOn3_Build_Ribozyme"
TutorialDir="/scratch/$USER/TutorialData/HandsOn3_Build_Ribozyme"
mkdir -p $TutorialDir
cp -r $DataDir/* $TutorialDir
cd $TutorialDir
cp -r input working_dir
cd working_dir
Option B — Downloading the archive
Hands-On Session 3 build files archive (tar.gz)
The archive does not include a full copy of modXNA. That dependency
is cloned separately later in the tutorial.
flowchart LR
%% =========================
%% Clean structure
%% =========================
A1["Input structure<br/>
7v9e.pdb"]
P1{{"<b>[§3.2.2]</b>
pdb4amber"}}
O1["<b>[from §3.2.2]</b>
Cleaned PDB<br/>7v9e_4amber.pdb"]
A1 --> P1 --> O1
%% =========================
%% Build custom parameters (modXNA)
%% =========================
A2["Input files<br/>
in-N3C.modxna<br/>modXNA/dat fragments"]
P2{{"<b>[§3.2.3.1]</b>
modxna.sh"}}
O2["<b>[From §3.2.3.1]</b>
C10 parameter file<br/>OKC.lib"]
A2 --> P2 --> O2
%% =========================
%% Build custom parameters (RESP)
%% =========================
A3["Input files<br/>
GUN.pdb<br/>"]
P3_1{{"<b>[§3.2.6.1]</b>
Gaussian"}}
A3 --> P3_1
P3_1 --> O3_1
O3_1["<b>[From §3.2.6.1]</b>
GUN.gjf<br/>"]
P3{{"<b>[§3.2.6.2]</b>
Gaussian"}}
O3["<b>[From §3.2.6.2]</b>
ESP output<br/>GUN.log"]
P4{{"<b>[§3.2.6.3]</b>
Antechamber<br/>"}}
O4["<b>[From §3.2.6.3]</b>
GUN.mol2"]
O3_1 --> P3 --> O3 --> P4 --> O4
P5{{"<b>[§3.2.6.4]</b>
tleap"}}
O5_1["<b>[From §3.2.6.4]</b>
GUN.lib"]
O5_2["<b>[From §3.2.6.4]</b>
GUN.parm7<br/>GUN.rst7"]
O4 --> P5
P5 --> O5_1
P5 --> O5_2
P6{{"<b>[§3.2.6.4]</b>
ParmEd"}}
O6["<b>[From §3.2.6.4]</b>
GUN.frcmod"]
O5_2 --> P6
P6 --> O6
%% =========================
%% Prepare build PDB
%% =========================
P7{{"<b>[§3.2.3.4]</b>
Manual Editing Structures<br/>
copy and edit PDB<br/>OKC/GUN/MG cleanup"}}
O7["<b>[From §3.2.3.4]</b>
Edited build PDB - 7v9e_prot.pdb"]
O1 --> P7
O2 --> P7
O5_1 --> P7
O6 --> P7
P7 --> O7
%% =========================
%% No-solvent build with 7v9e_prot.pdb
%% =========================
A4["<b>[From §3.2.3.1]</b>
<b>[From §3.2.6.4]</b>
Parameter and force-field inputs - <br/>OKC.lib<br/>GUN.lib<br/>GUN.frcmod<br/>modXNA/dat/frcmod.modxna"]
P8{{"<b>[§3.2.4.1]</b>
tleap"}}
O8["<b>[From §3.2.4.1]</b>
No-solvent system<br/>MTR1_noSolvent.parm7<br/>MTR1_noSolvent.rst7"]
O7 --> P8
A4 --> P8
P8 --> O8
%% =========================
%% Fix 5 prime terminus for later builds
%% =========================
P9{{"<b>[§3.2.4.1]</b>
Manual Editing<br/>remove 5 prime phosphate"}}
O9["<b>[From §3.2.4.1]</b>
Fixed PDB<br/> 7v9e_C5.pdb"]
O7 --> P9
P9 --> O9
%% =========================
%% Neutralized build
%% =========================
A5["<b>[From §3.2.3.1]</b>
<b>[From §3.2.6.4]</b>
Parameter and force-field inputs<br/>OKC.lib<br/>GUN.lib<br/>GUN.frcmod<br/>modXNA/dat/frcmod.modxna"]
P10{{"<b>[§3.2.4.2]</b>
tleap"}}
O10["<b>[From §3.2.4.2]</b>
Neutralized system - <br/>MTR1_counterIon.parm7<br/>MTR1_counterIon.rst7"]
O9 --> P10
A5 --> P10
P10 --> O10
%% =========================
%% Solvated build
%% =========================
P11{{"<b>[§3.2.4.3]</b>
tleap"}}
O11_1["<b>[From §3.2.4.3]</b>
Solvated topology<br/>MTR1_counterIon_water.parm7"]
O11_2["<b>[From §3.2.4.3]</b>
Solvated coordinates<br/>MTR1_counterIon_water.rst7"]
O9 --> P11
A5 --> P11
P11 --> O11_1
P11 --> O11_2
%% =========================
%% Bulk-ion count and build
%% =========================
A6["Input file<br/>
parmutils-GetNumIons.py"]
P12{{"<b>[§3.2.4.4]</b>
Parmed"}}
O12["<b>[From §3.2.4.4]</b>
Bulk-ion count<br/>41 Na<br/>41 Cl"]
O11_1 --> P12
A6 --> P12
P12 --> O12
P13{{"<b>[§3.2.4.4]</b>
tleap"}}
O13["<b>[From §3.2.4.4]</b>
Bulk-ion simulation box - <br/>MTR1_counterIon_water_bulk.parm7<br/>MTR1_counterIon_water_bulk.rst7<br/>MTR1_counterIon_water_bulk_amber.pdb"]
O9 --> P13
A5 --> P13
O12 --> P13
P13 --> O13
%% =========================
%% Apply 12-6-4 PGY corrections
%% =========================
A7["12-6-4 correction inputs<br/>
1264.sh<br/>parmed_1264_na.in<br/>lj_1264_pol.dat"]
P14{{"<b>[§3.2.5]</b>
ParmEd"}}
O14["<b>From [§3.2.5]</b>
12-6-4 corrected files - <br/>MTR1_counterIon_water_bulk_1264.parm7<br/>MTR1_counterIon_water_bulk_1264.rst7"]
O13 --> P14
A7 --> P14
P14 --> O14
%% =========================
%% Styling
%% =========================
classDef file fill:#fff7e6,stroke:#d98c00,stroke-width:1.5px,color:#111;
classDef program fill:#e8f1ff,stroke:#1f77b4,stroke-width:1.8px,color:#111;
classDef result fill:#eaf7ea,stroke:#2ca02c,stroke-width:1.5px,color:#111;
class A1,A2,A3,A4,A5,A6,A7 file;
class P1,P2,P3,P3_1,P4,P5,P6,P7,P8,P9,P10,P11,P12,P13,P14 program;
class O1,O2,O3,O3_1,O4,O5_1,O5_2,O6,O7,O8,O9,O10,O11_1,O11_2,O12,O13,O14 result;
3.2.2. Cleaning the PDB file
Work inside your dedicated tutorial directory. Treat
HandsOn3_Build_Ribozyme as read-only reference
material. If you are unable to produce an output for a given step, copy
the corresponding file from the reference output directory into your
working directory so that you can continue.
Set up your working directory once and copy the initial inputs into it:
mkdir -p MTR1_build
cd MTR1_build
cp -r ../../input/build/* ./
Unless stated otherwise, run all remaining commands in this tutorial
from MTR1_build.
The crystal structure of MTR1 can be obtained from the Protein Data Bank under the ID 7v9e.
Download the structure in PDB file format.
Note
At the workshop, 7v9e.pdb is already available in the provided files
on the DIPC cluster — no download needed.
Figure 1. PDB webpage where 7v9e.pdb can be downloaded.
Move this file to your working directory.
You have been provided a set of representations to visualize various
aspects of the crystal structure in MTR_crystalreps.txt:
# MTR1 crystal representations
display depthcue off
mol showrep top 0 on
mol selection {all}
mol representation Lines
mol addrep top
# Ions and water (vmd thinks the ligand is solvent, so exclude it)
mol selection {solvent and (not resname GUN)}
mol representation CPK
mol addrep top
# Active site residues
mol selection {resid 10 45 63 101}
mol representation licorice
mol addrep top
Open the pdb file in vmd:
vmd 7v9e.pdb
Source the VMD representation file MTR_crystalreps.txt. You can do
this from the Tk Console, or directly when launching VMD with
vmd -e MTR_crystalreps.txt 7v9e.pdb.
In the VMD main window:
Open the Extensions menu
Select Tk Console
In the Tk Console, type:
source MTR_crystalreps.txt
You will see various crystallographic waters and ions that we will ultimately clean from the pdb file before building the system. You will also see that the ribozyme contains two loops and a stem region that meet at a 3 way junction. Junctions are important in RNA structures because they involve bringing the negatively charged phosphates from the backbone close together. This is typically mediated by essential interactions with solvent. Notice that the active site, shown in licorice, is located at the junction.
Hide the rest of the RNA and focus on the active site. To do so, in the Graphical Representations window, double click on all and solvent and (not resname GUN):
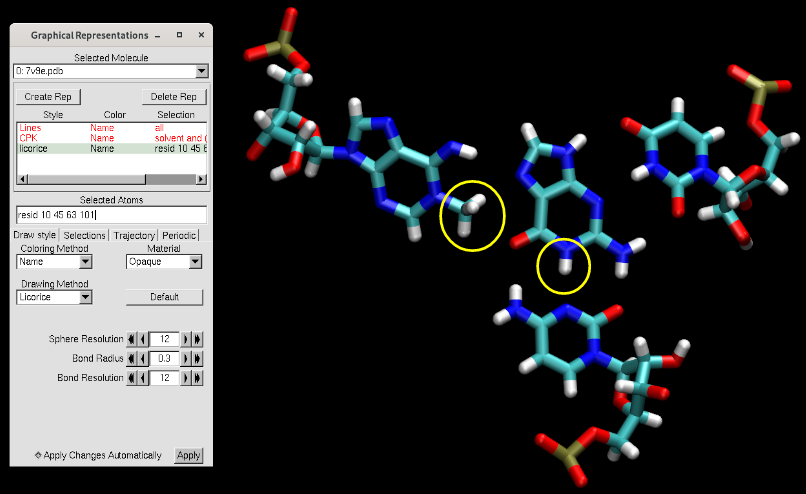
Figure 2. Active site of the MTR1 ribozyme from the crystal structure (PDB ID 7v9e).
This crystal captures the product state of the ribozyme. We see that adenine 63 is methylated at the N1 position. The ribozyme binds O6 methylated guanine to initiate the reaction, so the reactant state would have the methyl group on guanine and a standard adenine for 63. While in the product state N1 of guanine is protonated, the reactant state O6 methylated guanine does not have a proton at N1. We will hypothesize the source of that proton to be N3 position of C10, meaning the reaction starts with C10 protonated with a +1 charge.
Return to your terminal.
PDB files contain many lines giving information about the structure, as well as coordinate information for each resolved atom. More information regarding pdb format can be found here. The flags corresponding to atoms and their coordinates, which are the most relevant information for MD simulations, are “ATOM” and “HETATM” where ATOM corresponds main coordinate of the atom, HETATM corresponds to coordinates of solvent and ligand atoms not part of the main structure.
Take a look at the PDB file in vim and scroll through to get a sense of the contents.
This pdb file has anisotropic temperature factors reported in ANISOU lines. Since we will have to manually edit some lines in our pdb, we want to work with the minimalist version of the file. We will use the pdb4amber program to achieve that.
Make sure the Amber module is loaded, for example:
module load Amber/25.0-foss-2022a-AmberTools-25-CUDA-12.1.1-DeePMD
Run pdb4amber with 7v9e.pdb as input to generate 7v9e_4amber.pdb:
pdb4amber --in 7v9e.pdb --out 7v9e_4amber.pdb
Expected output:
==================================================
Summary of pdb4amber for: 7v9e.pdb
===================================================
----------Chains
The following (original) chains have been found:
A
---------- Alternate Locations (Original Residues!))
The following residues had alternate locations:
None
-----------Non-standard-resnames
GUN
---------- Gaps (Renumbered Residues!)
gap of 8.021418 A between G 61 and G 63
---------- Missing heavy atom(s)
None
Compare the two pdb files in vmd:
vmd -m 7v9e.pdb 7v9e_4amber.pdb
Turn each molecule on and off, to do so, click on “D” (i.e display) on the VMD Main window.
Check that they have the same number of atoms.
The chemically relevant structure should be the same, but the two files
are not strictly identical. pdb4amber removed introductory lines,
ANISOU lines and CONECT lines at the end, and renumbered atoms and
residues to be sequential (i.e. residue and/or atom numbers might be
different between the two files). Residue 62
(previously 63) is the 1-methyl adenosine, which was not recognized as a
standard RNA residue, causing a perceived gap between G 61 and G 63.
This is not a problem. Next we will make some manual modifications to
our pdb, so let’s first make a new copy to work with.
Copy 7v9e_4amber.pdb to 7v9e_prot.pdb
cp 7v9e_4amber.pdb 7v9e_prot.pdb
Open 7v9e_prot.pdb in vim.
The remaining REMARK lines would be important for other types of MD simulations such as crystal simulations, but won’t be relevant in this exercise. So if desired they can be removed, but also keeping them will have no effect. The following text blocks have line numbers turned on in vim (:set nu) to help you navigate the file.
Optional: Delete the REMARK lines at the top of the file:
REMARK 290 SMTRY1 1 1.000000 0.000000 0.000000 0.00000
REMARK 290 SMTRY2 1 0.000000 1.000000 0.000000 0.00000
REMARK 290 SMTRY3 1 0.000000 0.000000 1.000000 0.00000
REMARK 290 SMTRY1 2 -1.000000 0.000000 0.000000 0.00000
REMARK 290 SMTRY2 2 0.000000 -1.000000 0.000000 0.00000
REMARK 290 SMTRY3 2 0.000000 0.000000 1.000000 49.96950
REMARK 290 SMTRY1 3 -1.000000 0.000000 0.000000 0.00000
REMARK 290 SMTRY2 3 0.000000 1.000000 0.000000 0.00000
REMARK 290 SMTRY3 3 0.000000 0.000000 -1.000000 49.96950
REMARK 290 SMTRY1 4 1.000000 0.000000 0.000000 0.00000
REMARK 290 SMTRY2 4 0.000000 -1.000000 0.000000 0.00000
REMARK 290 SMTRY3 4 0.000000 0.000000 -1.000000 0.00000
3.2.3. Parametrization of non-standard residues
When working with modified nucleic acids in Amber, non-standard residues are often not available in standard force fields. In this activity, we will primarily focus on the modXNA workflow for parametrizing modified nucleotides. In particular, a protonated cytosine will be modeled as a non-standard nucleotide generated using modXNA. We will also provide the parameters for O6-methylguanine (O6mG) obtained using the classical RESP/Antechamber approach. An additional optional exercise describing this traditional parametrization procedure is proposed, but it will be covered in detail later in a separate activity dedicated to ligand parametrization.
3.2.3.1. N3-protonated cytidine using modXNA
We would like to “protonate” C10. Since a protonated cytosine is not a standard residue, we will use external parameters provided by modXNA. Indeed, modXNA provides a modular, fragment-based framework for building modified nucleotides compatible with modern Amber nucleic acid force fields [4].
3.2.3.1.1. Obtaining modXNA
The workshop archive provides the in-N3C.modxna input file, but not
the full modXNA program. Clone the complete modXNA repository
separately so that the script and fragment library are both available.
From inside MTR1_build, run:
git clone https://github.com/drroe/modXNA.git
If the modXNA directory already exists in MTR1_build, do not
clone again into the same location. Instead, reuse that existing
directory.
This directory contains:
modxna.sh— the main driver scriptdat/— the fragment library (backbone, sugar and base modules)
You should run modxna.sh from within this directory so that the
dat/ folder is found automatically.
modXNA requires AmberTools 25. In particular, the following
AmberTools components are used internally:
cpptrajtleap
Make sure the Amber module is loaded before running modXNA. For example:
module load Amber/25.0-foss-2022a-AmberTools-25-CUDA-12.1.1-DeePMD
3.2.3.1.2. modXNA input file
modXNA requires an input text file that specifies which fragments should be assembled to create a modified nucleotide.
Each line of the file follows the format:
<backbone ID> <sugar ID> <base ID>
3.2.3.1.3. Fragment selection
For this example, we build an RNA residue containing a protonated N3 cytidine. The required fragments are:
RNA backbone:
RPO(standard dimethyl phosphate backbone, charge −1, atom types from OL3)RNA sugar:
RC3(standard ribose sugar in C3′-endo conformation, atom types from OL3)Modified base:
N3C(protonated cytidine at N3, charge +1)
Fragment codes can be checked in the modXNA fragment catalog: https://modxna.chpc.utah.edu/catalog/
3.2.3.1.4. Creating the input file
The file in-N3C.modxna is already included in
HandsOn3_Build_Ribozyme/input/build and was copied
into MTR1_build during setup. Its content is:
### RNA residue with protonated N3 of cytidine
RPO RC3 N3C
3.2.3.1.5. Running modXNA
Run modXNA using the input file:
cd modXNA
chmod +x modxna.sh
./modxna.sh -i ../modXNA-files/in-N3C.modxna
3.2.3.1.6. What modXNA does
The program will:
locate the requested fragments in the
dat/directory,remove the capping groups from each fragment,
assemble the backbone, sugar, and base into a single nucleotide,
adjust partial charges to ensure consistency,
perform a short energy minimization to optimize the resulting structure.
When modXNA is run, it automatically assigns a random three-letter name to the newly generated residue. In our example, the residue is named OKC. modXNA produces an OKC.lib file, which can be used to build Amber topology and coordinate files for molecular dynamics simulations.
To incorporate our new residue into our PDB file, rename residue 9
(which corresponds to C10 in the original pdb file before renumbering)
from C to OKC in 7v9e_prot.pdb and save the file.
In vim, use Visual Block mode to edit all lines of residue 9 at once:
Open the file and search for the first atom of residue 9:
/C A 9Position the cursor on the
Cin column 18 (the residue name field).Press
Ctrl+Vto enter Visual Block mode.Press
jrepeatedly to extend the selection down through all atom lines of residue 9.Press
llto extend the selection to cover the full 3-character residue name field.Press
c, typeOKC, then pressEsc. All selected lines are updated at once.Save with
:wq.
Warning
When editing PDB files, it is critical to preserve the fixed-column formatting of the PDB standard. Each field must remain within its designated column range; otherwise, errors may occur when building the system in LEaP.
Hint
The PDB format uses fixed-width columns. Each field must remain within its designated column range; otherwise, errors may occur when building the system in LEaP.
Columns |
Column description |
|---|---|
1-6 |
Indicates the |
7-11 |
Atom serial number |
13-16 |
Atom name |
17 |
Alternate location indicator |
18-20 |
Residue name |
22 |
Chain identifier |
23-26 |
Residue sequence number |
27 |
Code for residue insertion |
31-38 |
Orthogonal coordinates for X (Angstrom) |
39-46 |
Orthogonal coordinates for Y (Angstrom) |
47-54 |
Orthogonal coordinates for Z (Angstrom) |
55-60 |
Occupancy |
61-66 |
Temperature factor (B-factor) |
77-78 |
Element symbol |
79-80 |
Atom charge |
ATOM 262 P OKC A 9 18.771 0.638 43.621 1.00 55.52 P
ATOM 263 OP1 OKC A 9 19.026 -0.826 43.656 1.00 74.66 O
ATOM 264 OP2 OKC A 9 19.830 1.583 44.056 1.00 70.25 O
ATOM 265 O5' OKC A 9 18.387 1.029 42.130 1.00 55.31 O
ATOM 266 C5' OKC A 9 17.258 0.451 41.499 1.00 47.08 C
ATOM 267 C4' OKC A 9 16.859 1.235 40.281 1.00 48.52 C
ATOM 268 O4' OKC A 9 16.281 2.504 40.665 1.00 44.83 O
ATOM 269 C3' OKC A 9 18.003 1.628 39.361 1.00 44.20 C
ATOM 270 O3' OKC A 9 18.443 0.567 38.545 1.00 42.92 O
ATOM 271 C2' OKC A 9 17.417 2.801 38.599 1.00 45.04 C
ATOM 272 O2' OKC A 9 16.550 2.358 37.563 1.00 50.64 O
ATOM 273 C1' OKC A 9 16.599 3.485 39.695 1.00 46.71 C
ATOM 274 N1 OKC A 9 17.356 4.568 40.353 1.00 47.41 N
ATOM 275 C2 OKC A 9 17.526 5.751 39.645 1.00 47.12 C
ATOM 276 O2 OKC A 9 17.052 5.815 38.499 1.00 46.70 O
ATOM 277 N3 OKC A 9 18.205 6.764 40.225 1.00 43.87 N
ATOM 278 C4 OKC A 9 18.701 6.624 41.456 1.00 46.67 C
ATOM 279 N4 OKC A 9 19.363 7.668 41.963 1.00 39.22 N
ATOM 280 C5 OKC A 9 18.528 5.423 42.206 1.00 49.00 C
ATOM 281 C6 OKC A 9 17.859 4.425 41.614 1.00 49.69 C
ATOM 282 H5' OKC A 9 16.517 0.434 42.124 1.00 56.60 H
ATOM 283 H5'' OKC A 9 17.470 -0.459 41.237 1.00 56.60 H
ATOM 284 H4' OKC A 9 16.194 0.731 39.786 1.00 58.32 H
ATOM 285 H3' OKC A 9 18.752 1.941 39.891 1.00 53.14 H
ATOM 286 H2' OKC A 9 18.103 3.391 38.250 1.00 54.15 H
ATOM 287 HO2' OKC A 9 16.855 2.616 36.823 1.00 60.87 H
ATOM 288 H1' OKC A 9 15.781 3.844 39.317 1.00 56.16 H
ATOM 289 H41 OKC A 9 19.699 7.621 42.753 1.00 47.16 H
ATOM 290 H42 OKC A 9 19.452 8.387 41.500 1.00 47.16 H
ATOM 291 H5 OKC A 9 18.866 5.336 43.069 1.00 58.90 H
ATOM 292 H6 OKC A 9 17.735 3.623 42.068 1.00 59.73 H
3.2.3.2. From the methylated adenine to a standard one
Next we will “mutate” the methylated adenine, 1MA, back to a
standard adenine. This requires editing 7v9e_prot.pdb, changing the
residue name from 1MA to A, and deleting the methyl atoms. We
will start by deleting the methyl atoms so there are fewer lines to edit.
Delete the methyl atoms and additional hydrogen from residue 62 (A63 in
the original pdb file before renumbering): CM1, HN61, HM11,
HM12, and HM13.
Change the residue name of 62 from 1MA to A, by changing 1 to A, and
replacing M and A with two spaces, so that the total number of
columns is not modified.
Use Visual Block mode in vim to edit all lines at once:
Search for the first atom of residue 62:
/1MAPosition the cursor on the
1in column 18.Press
Ctrl+Vto enter Visual Block mode.Press
jto extend down through all remaining1MAatom lines.Press
llto select the 3-character residue name field.Press
c, typeA `` (A followed by two spaces), then press ``Esc.Save with
:wq.
ATOM 1946 P A A 62 22.724 23.209 43.994 1.00 70.56 P
ATOM 1947 OP1 A A 62 23.291 23.505 45.361 1.00 73.73 O
ATOM 1948 OP2 A A 62 23.176 24.033 42.821 1.00 88.65 O
ATOM 1949 O5' A A 62 22.969 21.684 43.646 1.00 63.06 O
ATOM 1950 C5' A A 62 24.006 20.974 44.301 1.00 68.95 C
ATOM 1951 C4' A A 62 23.489 20.121 45.431 1.00 58.78 C
ATOM 1952 O4' A A 62 22.595 19.115 44.876 1.00 60.31 O
ATOM 1953 C3' A A 62 24.569 19.343 46.178 1.00 56.81 C
ATOM 1954 O3' A A 62 24.128 19.101 47.511 1.00 55.15 O
ATOM 1955 C2' A A 62 24.610 18.029 45.411 1.00 51.57 C
ATOM 1956 O2' A A 62 25.177 16.949 46.126 1.00 52.34 O
ATOM 1957 C1' A A 62 23.117 17.826 45.125 1.00 54.19 C
ATOM 1958 N9 A A 62 22.827 16.957 43.975 1.00 54.44 N
ATOM 1959 C8 A A 62 23.376 17.668 42.954 1.00 52.77 C
ATOM 1960 N7 A A 62 23.192 16.958 41.807 1.00 40.69 N
ATOM 1961 C5 A A 62 22.523 15.840 42.134 1.00 45.39 C
ATOM 1962 C6 A A 62 22.012 14.634 41.321 1.00 49.52 C
ATOM 1963 N6 A A 62 22.234 14.614 40.078 1.00 44.46 N
ATOM 1964 N1 A A 62 21.313 13.559 41.983 1.00 45.82 N
ATOM 1966 C2 A A 62 21.087 13.586 43.394 1.00 45.05 C
ATOM 1967 N3 A A 62 21.577 14.723 44.151 1.00 48.62 N
ATOM 1968 C4 A A 62 22.304 15.843 43.481 1.00 49.84 C
ATOM 1969 H5' A A 62 24.644 21.612 44.658 1.00 82.84 H
ATOM 1970 H5'' A A 62 24.449 20.402 43.655 1.00 82.84 H
ATOM 1971 H4' A A 62 22.998 20.672 46.060 1.00 70.64 H
ATOM 1972 H3' A A 62 25.426 19.797 46.154 1.00 68.27 H
ATOM 1973 H2' A A 62 25.087 18.142 44.574 1.00 61.99 H
ATOM 1974 HO2' A A 62 25.201 17.142 46.957 1.00 62.91 H
ATOM 1975 H1' A A 62 22.686 17.458 45.912 1.00 65.13 H
ATOM 1976 H8 A A 62 23.815 18.352 42.502 1.00 63.43 H
ATOM 1981 H2 A A 62 20.636 12.891 43.817 1.00 54.16 H
3.2.3.3. Using the provided ligand parameters for O6 methylguanine (O6mG)
The ligand in this system is O6-methylguanine, with residue name
GUN. Although O6-methylated guanine is available in modXNA, that
implementation assumes the presence of a nucleic-acid backbone and
sugar. In our case, the ligand consists only of the base capped with a
hydrogen atom, so it is treated as a separate non-standard residue.
For the main workflow of this tutorial, the required ligand parameter files are already provided in the workshop archive:
GUN.libGUN.frcmod
You do not need to derive these parameters in order to complete the
system-building workflow. The derivation of GUN parameters from
Gaussian, RESP, antechamber, and parmed is moved to an optional
exercise at the end of this tutorial.
The remaining task for the main workflow is to make sure that the atom
names in 7v9e_prot.pdb are consistent with the provided GUN.lib
file before building the final system.
3.2.3.4. Finalization of system preparation
The atom names reported in the pdb are different than atom names we have in our parameter file.
Print the atom types in GUN.lib:
head -n22 GUN.lib
Expected output:
!!index array str
"GUN"
!entry.GUN.unit.atoms table str name str type int typex int resx int flags int seq int elmnt dbl chg
"N1" "na" 0 1 131072 1 7 -0.334523
"C1" "cc" 0 1 131072 2 6 0.097034
"N2" "nd" 0 1 131072 3 7 -0.523097
"C2" "ca" 0 1 131072 4 6 0.144003
"C3" "ca" 0 1 131072 5 6 0.558035
"O1" "os" 0 1 131072 6 8 -0.329698
"N3" "nb" 0 1 131072 7 7 -0.745822
"C4" "ca" 0 1 131072 8 6 0.884237
"N4" "nh" 0 1 131072 9 7 -0.890772
"N5" "nb" 0 1 131072 10 7 -0.693379
"C5" "ca" 0 1 131072 11 6 0.325075
"H1" "hn" 0 1 131072 12 1 0.347262
"H2" "h5" 0 1 131072 13 1 0.176097
"H3" "hn" 0 1 131072 14 1 0.381855
"H4" "hn" 0 1 131072 15 1 0.381855
"C6" "c3" 0 1 131072 16 6 -0.005320
"H5" "h1" 0 1 131072 17 1 0.075719
"H6" "h1" 0 1 131072 18 1 0.075719
"H7" "h1" 0 1 131072 19 1 0.075719
We need to rename the heavy atoms in the pdb file 7v9e_prot.pdb to their corresponding
names in the parameter file. In order to simplify this process we will
first remove the protons within the residue. LEaP can easily add these
back later with the correct names.
Delete the protons HN1, HN9, H8, HN21, and HN22.
Rename the heavy atoms as follows:
HETATM 2175 N1 GUN A 69 20.435 11.931 36.172 1.00 40.72 N
HETATM 2176 C1 GUN A 69 21.059 12.713 37.080 1.00 45.85 C
HETATM 2177 N2 GUN A 69 21.000 12.094 38.277 1.00 37.65 N
HETATM 2178 C2 GUN A 69 20.352 10.944 38.115 1.00 42.37 C
HETATM 2179 C3 GUN A 69 19.965 9.819 39.086 1.00 43.44 C
HETATM 2180 O1 GUN A 69 20.255 9.900 40.233 1.00 39.68 O
HETATM 2181 N3 GUN A 69 19.246 8.676 38.614 1.00 43.96 N
HETATM 2182 C4 GUN A 69 18.873 8.588 37.236 1.00 43.78 C
HETATM 2183 N4 GUN A 69 18.150 7.445 36.734 1.00 40.36 N
HETATM 2184 N5 GUN A 69 19.248 9.652 36.333 1.00 41.19 N
HETATM 2185 C5 GUN A 69 19.996 10.845 36.806 1.00 45.72 C
Barium and sodium ions, together with a small number of water molecules, were resolved crystallographically. These species are not catalytically relevant. For this reason, sodium ions and crystallographic water molecules were removed. The barium ions were intentionally retained and subsequently replaced by magnesium ions in order to illustrate in practice how 12-6-4 Lennard-Jones correction terms with Panteva–Giambasu–York (PGY) pairwise corrections are applied, enabling balanced interactions with nucleic acids.
Delete sodium ion and water molecules.
(In vim: :g/HETATM.*\(NA\|HOH\)/d to delete all sodium and water HETATM lines.)
HETATM 2200 NA NA A 79 36.287 16.215 43.272 1.00 34.26 NA
HETATM 2201 NA NA A 80 31.896 7.690 34.088 1.00 50.91 NA
HETATM 2202 O HOH A 81 43.549 22.559 52.718 1.00 67.88 O
HETATM 2203 O HOH A 82 46.224 26.856 56.524 1.00 69.56 O
HETATM 2204 O HOH A 83 -0.607 23.932 34.220 1.00 62.80 O
HETATM 2205 O HOH A 84 13.884 27.718 35.721 1.00 64.85 O
HETATM 2206 O HOH A 85 15.463 4.299 36.801 1.00 47.72 O
HETATM 2207 O HOH A 86 8.577 12.465 25.488 1.00 70.15 O
HETATM 2208 O HOH A 87 27.735 18.894 46.490 1.00 40.64 O
HETATM 2209 O HOH A 88 33.130 -4.678 33.799 1.00 32.49 O
HETATM 2210 O HOH A 89 32.100 4.191 32.161 1.00 39.50 O
HETATM 2211 O HOH A 90 26.286 0.200 32.199 1.00 50.23 O
HETATM 2212 O HOH A 91 36.070 10.612 25.896 1.00 55.39 O
HETATM 2213 O HOH A 92 25.235 17.366 32.641 1.00 57.43 O
HETATM 2214 O HOH A 93 34.079 -14.419 28.448 1.00 52.99 O
HETATM 2215 O HOH A 94 35.911 -4.317 32.231 1.00 33.16 O
HETATM 2216 O HOH A 95 28.622 -6.340 30.512 1.00 55.39 O
HETATM 2217 O HOH A 96 21.541 18.363 31.228 1.00 45.76 O
HETATM 2218 O HOH A 97 39.960 0.108 40.497 1.00 46.79 O
HETATM 2219 O HOH A 98 5.321 31.207 42.190 1.00 59.35 O
HETATM 2220 O HOH A 99 -0.444 19.583 36.126 1.00 52.16 O
HETATM 2221 O HOH A 100 28.698 0.541 51.368 1.00 59.76 O
HETATM 2222 O HOH A 101 15.735 -0.430 36.539 1.00 55.39 O
HETATM 2223 O HOH A 102 37.524 9.301 27.209 1.00 62.05 O
HETATM 2224 O HOH A 103 28.598 21.370 47.356 1.00 48.57 O
HETATM 2225 O HOH A 104 33.224 6.071 32.685 1.00 44.69 O
HETATM 2226 O HOH A 105 28.951 21.487 44.742 1.00 56.37 O
HETATM 2227 O HOH A 106 30.511 21.905 41.786 1.00 63.69 O
HETATM 2228 O HOH A 107 33.481 24.017 45.493 1.00 47.89 O
HETATM 2229 O HOH A 108 -3.444 23.781 35.501 1.00 60.89 O
HETATM 2230 O HOH A 109 27.024 21.879 45.193 1.00 58.85 O
HETATM 2231 O HOH A 110 32.792 -2.728 32.364 1.00 43.28 O
HETATM 2232 O HOH A 111 30.418 23.355 48.435 1.00 55.87 O
HETATM 2233 O HOH A 112 31.672 27.219 56.474 1.00 56.58 O
And then, replace barium ions by magnesium ions.
(In vim: :%s/BA/MG/g to replace all occurrences of BA with MG.)
HETATM 2191 MG MG A 70 31.906 8.390 45.892 0.60 24.18 MG
HETATM 2192 MG MG A 71 34.813 -6.355 31.778 0.58 30.39 MG
HETATM 2193 MG MG A 72 31.033 0.308 49.682 0.40 61.24 MG
HETATM 2194 MG MG A 73 33.004 17.329 49.683 0.45 54.07 MG
HETATM 2195 MG MG A 74 27.824 -1.912 53.472 0.40 46.29 MG
HETATM 2196 MG MG A 75 32.282 24.572 53.839 0.48 69.02 MG
HETATM 2197 MG MG A 76 0.279 22.083 36.141 0.75 84.35 MG
HETATM 2198 MG MG A 77 18.560 14.938 32.414 0.56 62.27 MG
HETATM 2199 MG MG A 78 8.384 17.597 38.313 0.76120.11 MG
Save and close the pdb file.
Visually inspect 7v9e_prot.pdb in VMD to ensure you have made the
desired changes.
Source MTR_cleanreps.txt:
# MTR1 clean pdb representations
display depthcue off
mol showrep top 0 on
# Ions and water (vmd thinks the ligand is solvent, so exclude it)
mol selection {solvent and (not resname GUN)}
mol representation CPK
mol addrep top
# Active site residues
mol selection {resid 9 44 62 69}
mol representation licorice
mol addrep top
Figure 3. VMD representation of the MTR1 ribozyme after editing the PDB file.
There should be no solvent remaining, but you should still see the magnesium ions. Since the pdb file has been reordered, the active site residues have different numbers from the file we downloaded from the PDB website. The active site representation now contains residues 9, 44, 62, and 69, which used to be residues 10, 45, 63, and 101.
Source MTR_cleanreps.txt again to use the representations.
Hide all and focus on the active site in licorice.
Select some of the active site atoms to confirm that the residue names have been changed.
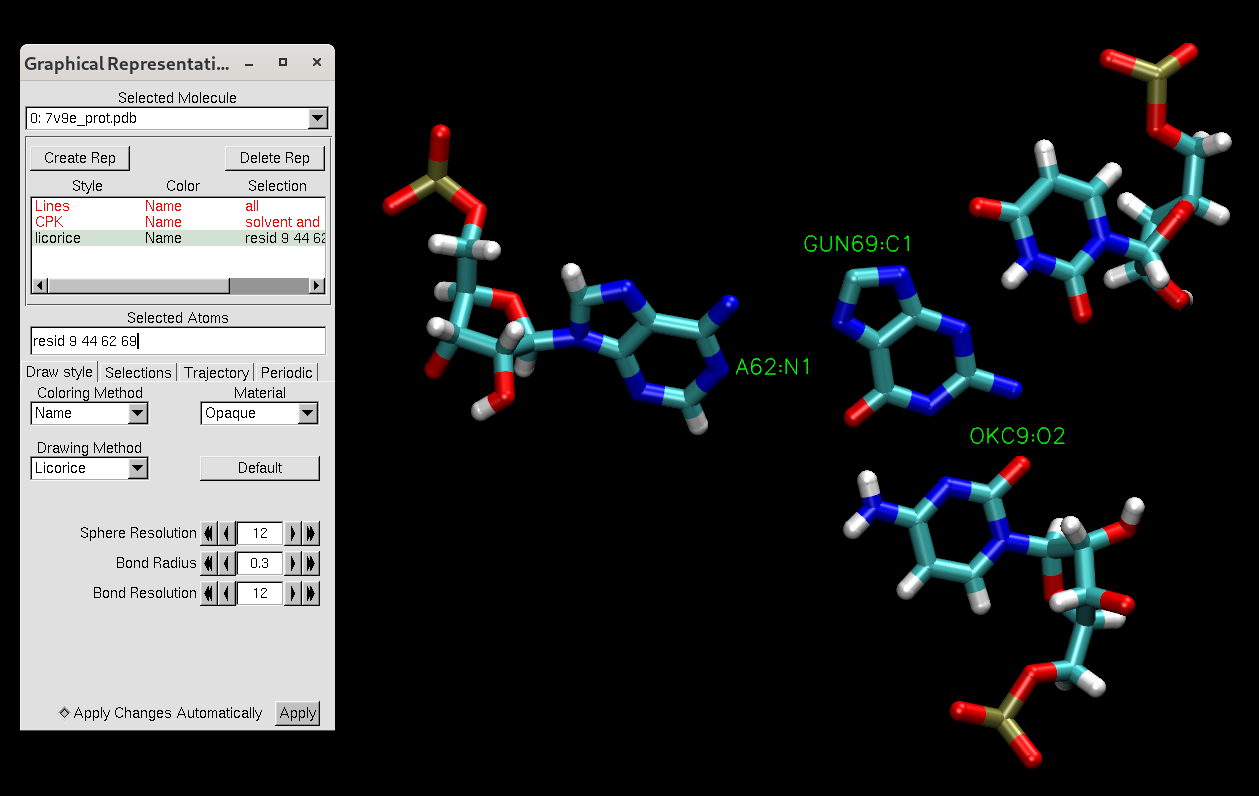
Figure 4. VMD representation of the MTR1 active site after editing the PDB file.
3.2.4. Building the Simulation Box
AMBER has a program named LEaP, which takes in coordinates and force field definitions and produces parameter and restart files needed for system definition by the MD engine. We will use LEaP to create a simulation box that has explicit water molecules, Na+, Cl- and Mg2+ ions, as well as our RNA-ligand system in its protonated state. This process is also referred to as “solvating the system”. LEaP can fill in missing atoms, but it will not delete atoms. This means the structure cannot contain atom types not included in a force field.
LEaP has a specific order of events. Force field parameters need to be loaded first, followed by loading the molecule. Next solvent and ions are added and finally output files need to be saved. Nucleic acids benefit from adding ions and water in a specific order. For pedagogical reasons, we will start with a step-wise approach to building the simulation box. At the end we will have a single input file that will build the simulation box in one go.
3.2.4.1. Force fields and loading the molecule
In order for a molecule to be loaded without error, every residue and atom in the input structure must have a corresponding parameter set already loaded in LEaP. AMBER provides a variety of well-established force fields for proteins and nucleic acids, as well as water models and metal ion parameters. The current recommendation for RNA simulations is the OL3 force field [5].
We use the TIP4P-Ew water model [6] for nucleic acid systems due to the availability of fine-tuned ion parameters compatible – which are often critical for nucleic acid structure and function – including 12-6-4 Lennard-Jones corrections for monovalent ions. For Mg2+ ions, Panteva–Giambasu–York (PGY) pairwise corrections are applied in conjunction with the 12-6-4 model to improve the description of RNA–magnesium interactions [7][8].
Additionally, our system contains two non-standard residues (i.e. residue names not readily available in the standard AMBER force fields), which have been newly parameterized. The corresponding parameter files are provided as OKC.lib, GUN.lib, and GUN.frcmod.
First, we verify that all required definitions are correctly loaded by building the topology and restart files for the RNA–ligand system alone, without any solvent.
Take a look at tleap_MTR1_param.in
set default nocenter on # Do not recenter the molecule
### Sourcing amber forcefields ###############################################
source leaprc.RNA.OL3 # Sourcing RNA force field OL3 to describe RNA.
source leaprc.water.tip4pew # Sourcing water model TIP4P-Ew
# and parameters for corresponding monovalent ions
loadamberparams frcmod.ions234lm_1264_tip4pew # divalent metal ion parameters
### Including non-standard residue parameters ################################
# Ligand O6 methylated guanine
loadoff GUN.lib
loadamberparams GUN.frcmod
# N3 protonated cytosine
loadoff OKC.lib
loadamberparams ./modXNA/dat/frcmod.modxna
### Loading structure from file ##############################################
mol= loadpdb "7v9e_prot.pdb"
### Saving output files #####################################################
# save parameter and restart files
saveamberparm mol MTR1_noSolvent.parm7 MTR1_noSolvent.rst7
quit
Run tleap with tleap_MTR1_param.in as input:
tleap -sf tleap_MTR1_param.in
The following error is left intentionally for pedagogical reasons so that you can see a common LEaP failure mode and fix it explicitly:
FATAL: Atom .R<C5 1>.A<P 30> does not have a type.
FATAL: Atom .R<C5 1>.A<OP1 31> does not have a type.
FATAL: Atom .R<C5 1>.A<OP2 32> does not have a type.
Warning
This error is expected at this stage of the tutorial. It is caused by a 5’ phosphate that is incompatible with the residue definition being used in LEaP. Do not ignore it. Fix the structure before proceeding.
These errors indicate that phosphate atoms are present at the 5’ end of the nucleic acid chain, which is incompatible with AMBER’s residue definitions. In AMBER, 5’ and 3’ terminal residues are treated differently: the 5’ end is capped at the O5’ atom, and this capping requires the absence of phosphate groups.
To fix this problem, the phosphate group must be removed from the 5’ terminal residue (residue 1). Since this involves modifying the structure file, we first create a copy of the original PDB file to preserve it.
Copy the file 7v9e_prot.pdb to a new file named 7v9e_C5.pdb. All
subsequent modifications to correct the error will be performed on this new
file.
cp 7v9e_prot.pdb 7v9e_C5.pdb
Delete the phosphate atoms (P, OP1, and OP2) from residue 1 in
7v9e_C5.pdb, as shown below:
ATOM 1 P C A 1 36.782 21.325 44.826 1.00 96.64 P
ATOM 2 OP1 C A 1 36.892 20.457 43.625 1.00 89.80 O
ATOM 3 OP2 C A 1 35.628 21.157 45.749 1.00 78.55 O
Then, you can define a new input file for tleap, with this new pdb.
Take a look at tleap_MTR1_param_C5.in
set default nocenter on # Do not recenter the molecule
### Sourcing amber forcefields ###############################################
source leaprc.RNA.OL3 # Sourcing RNA force field OL3 to describe RNA.
source leaprc.water.tip4pew # Sourcing water model TIP4P-Ew
# and parameters for corresponding monovalent ions
loadamberparams frcmod.ions234lm_1264_tip4pew # divalent metal ion parameters
### Including non-standard residue parameters ################################
# Ligand O6 methylated guanine
loadoff GUN.lib
loadamberparams GUN.frcmod
# N3 protonated cytosine
loadoff OKC.lib
loadamberparams ./modXNA/dat/frcmod.modxna
### Loading structure from file ##############################################
mol= loadpdb "7v9e_C5.pdb" #UPDATED PDB
### Saving output files #####################################################
# save parameter and restart files
saveamberparm mol MTR1_noSolvent.parm7 MTR1_noSolvent.rst7
quit
Run tleap with tleap_MTR1_param_C5.in as input:
tleap -sf tleap_MTR1_param_C5.in
Visualize the resulting parameter and restart file in vmd. You may continue to use the MTR_cleanreps.txt until otherwise specified.
vmd MTR1_noSolvent.parm7 MTR1_noSolvent.rst7
Hide all and focus on the active site.
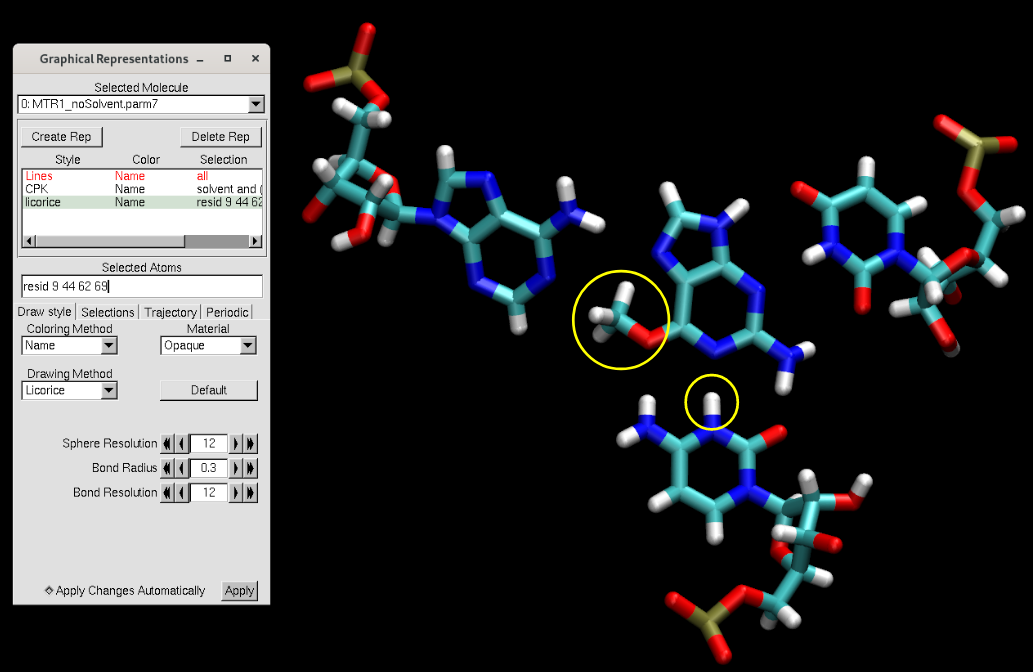
Figure 5. VMD representation of the MTR1 active site after adding missing atoms with LEaP.
You will see that the methyl group was added on the ligand, and a proton was added onto the cytosine (circled in yellow above). Additionally, missing hydrogen atoms were added.
3.2.4.2. Neutralize the system with counterions
Now we will only add the counter ions to our system. Use the tleap_MTR1_counterIon.in input file, which differs from the above by only the inclusion of the following line and output file names.
### Adding counter ions ######################################################
addions mol NA 0 # Add sodium until charge is zero
Run tleap with the following command:
tleap -f tleap_MTR1_counterIon.in
Visualize the output structures in vmd
vmd MTR1_counterIon.parm7 MTR1_counterIon.rst7
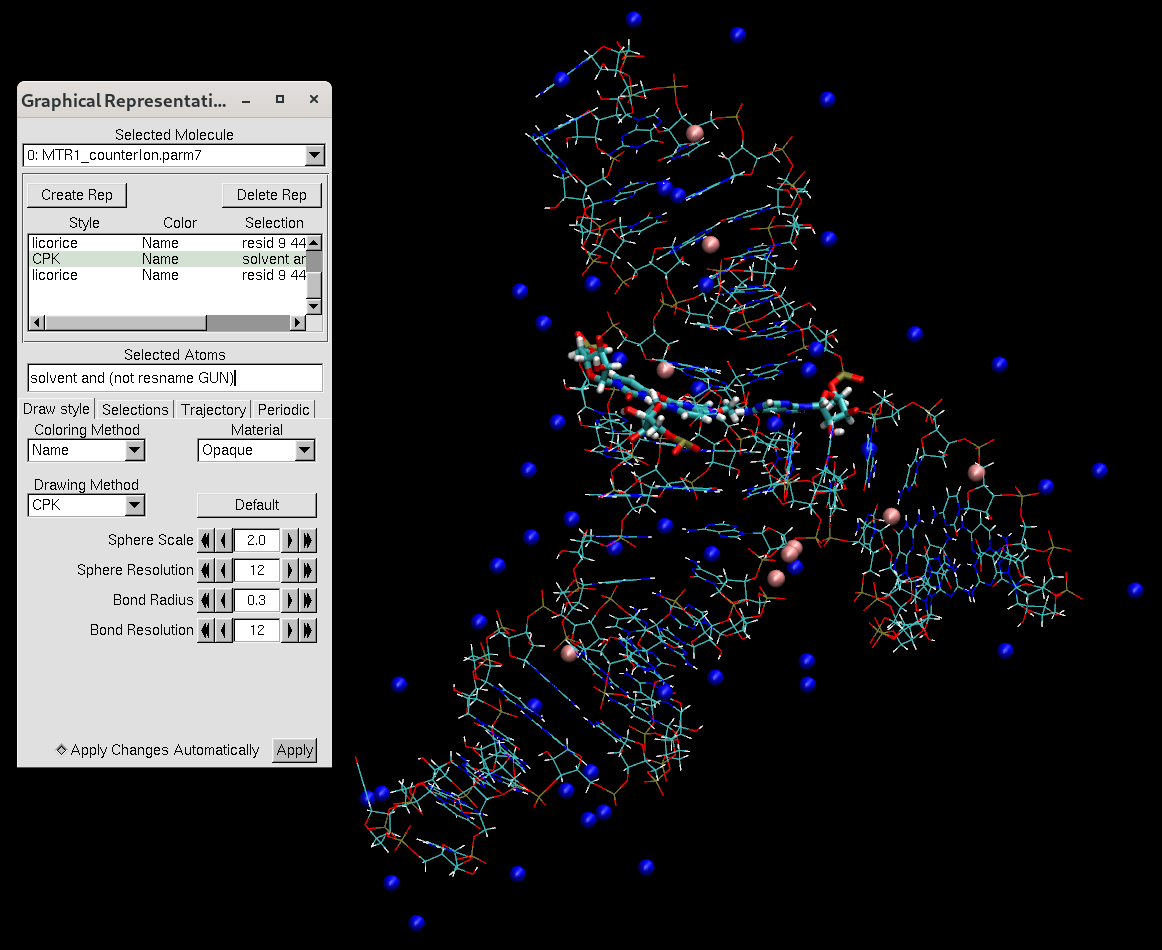
Figure 6. VMD representation of the MTR1 ribozyme after adding counter ions.
You will notice that the counterions are placed along the phosphate backbone, which is negatively charged. It is essential to neutralize the backbone to stabilize the structure. We add the counterions before adding water to avoid the ions being placed far away from the RNA.
3.2.4.3. Add water molecules
Next we will add water molecules. The simulations will ultimately take place under periodic boundary conditions, but we must define the shape of the box for a given image. Since the RNA is somewhat globular, we will choose a truncated octahedron. This is closer to a sphere than say, a rectangular box. The size of the box will define the volume of the system. We want to create a sufficient buffer of solvent around the RNA, so we will use a solvation radius of 9 Å. We will use a 4-point water model.
Now we will add the water to our system. Use the tleap_MTR1_counterIon_water.in input file, which differs from the above by only the inclusion of the following line and output file names.
### Defining box shape & size with chosen water model ########################
solvateoct mol TIP4PEWBOX 9 # solvate with truncated octahedron box 9A out
Run tleap with the following command:
tleap -f tleap_MTR1_counterIon_water.in
Visualize the structure in vmd using the MTR_waterreps.txt
representations:
vmd MTR1_counterIon_water.parm7 MTR1_counterIon_water.rst7
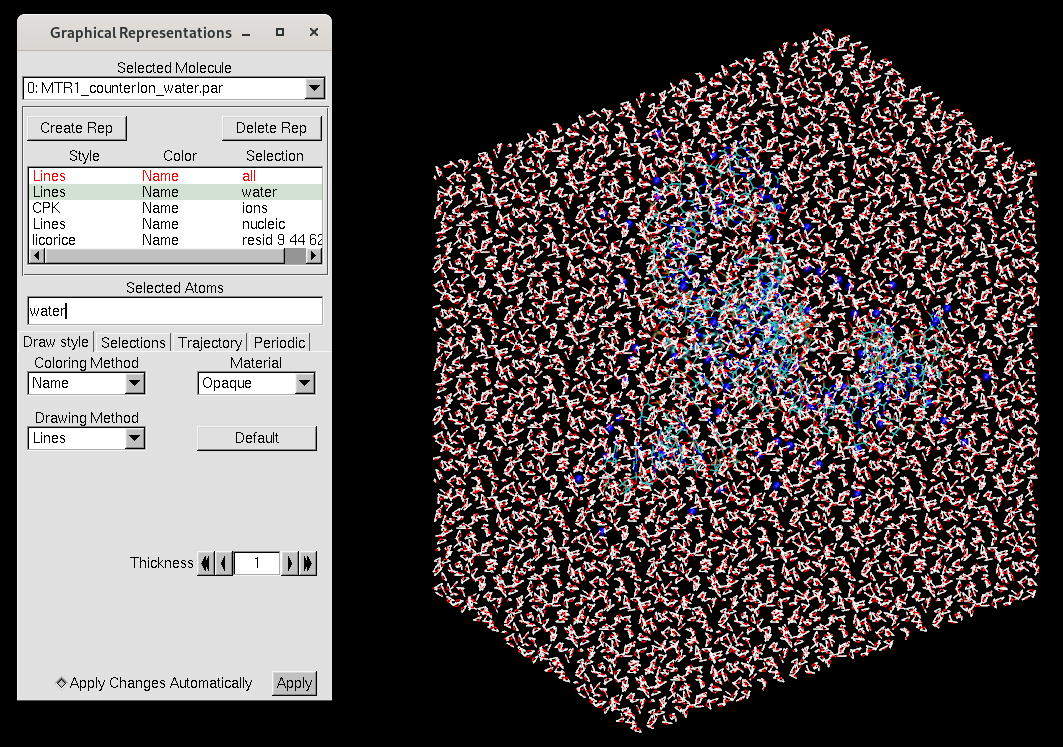
Figure 7. VMD representation of the MTR1 ribozyme in an octahedral solvation box.
The representations now separate the water and ions rather than referring to just solvent. Here are the contents of MTR_waterreps.txt:
# MTR1 solvated representations
display depthcue off
mol showrep top 0 off
# water
mol selection {water}
mol representation Lines
mol addrep top
# Ions (vmd thinks the ligand is solvent, so exclude it)
mol selection {ions}
mol representation CPK 2.0
mol addrep top
# RNA
mol selection {nucleic}
mol representation Lines
mol addrep top
# Active site residues
mol selection {resid 9 44 62 69}
mol representation licorice
mol addrep top
3.2.4.4. Add bulk ions
Next, bulk Na+ and Cl- ions must be added to the water box in order to reach a target concentration of 0.14 M, corresponding to physiological ionic concentration.
AMBER does not directly compute the number of counterions required to reach a desired bulk ion concentration. Therefore, this number must be calculated explicitly based on the system size, the number of water molecules, and the net charge of the system.
To perform this calculation, we use a helper script provided with the parameterization utilities:
parmutils-GetNumIons.py
This script analyzes an AMBER topology file and reports the number of ions that should be added in order to reach the desired salt concentration.
The content of the script is shown below for reference:
#!/usr/bin/env python3
import sys
import parmed
if len(sys.argv) < 2:
print("""
GetNumIons.py parm [conc]
parm amber parm7 filename (string)
conc ion concentration, M (float, optional, default: 0.14)
This script reports:
(1) the number of waters within the parm file
(2) the current counterion (Na/Cl) concentration
(3) the current net charge
(4) the net charge excluding Na/Cl counterions
The script provides the addions tleap-command that should be
used to add enough Na/Cl to neutralize the system and achieve
an excess counterion concentration of 0.14 M, or the
concentration specified by the optional argument.
""")
exit(0)
WatConc = 55. # M
IonConc = 0.14 # M
if len(sys.argv) > 2:
IonConc = float(sys.argv[2])
parm = parmed.load_file(sys.argv[1])
NumWatParm = 0
Charge = 0.
NonionCharge = 0.
NaParm = 0
ClParm = 0
for atom in parm:
Charge += atom.charge
NonionCharge += atom.charge
if atom.type == "OW" and atom.residue.name == "WAT":
NumWatParm += 1
elif atom.type == "Na+" or atom.type == "K+":
NaParm += 1
NonionCharge -= 1
elif atom.type == "Cl-":
ClParm += 1
NonionCharge += 1
ChargeFloat = Charge
NonionChargeFloat = NonionCharge
Charge = int("%.0f" % Charge)
NonionCharge = int("%.0f" % NonionCharge)
Na = ((NumWatParm + NaParm + ClParm) * IonConc / WatConc) \
- (NonionCharge if NonionCharge < 0 else 0) - NaParm
Cl = ((NumWatParm + NaParm + ClParm) * IonConc / WatConc) \
+ (NonionCharge if NonionCharge > 0 else 0) - ClParm
Na = int("%.0f" % Na)
Cl = int("%.0f" % Cl)
ParmConc = 0.
if NumWatParm > 0:
ParmConc = min(NaParm, ClParm) * WatConc / (NumWatParm + NaParm + ClParm)
print("Current ion concentration: %.3f M" % ParmConc)
print("Current non-counterion charge: %i %.6f" % (NonionCharge, NonionChargeFloat))
print("Current # WAT:", NumWatParm)
print("Current charge: %i %.6f" % (Charge, ChargeFloat))
if Na < 0 or Cl < 0:
print("ERROR: You already have too many counterions; don't add more")
if Na < 0:
print("ERROR: Remove %i sodium" % (-Na))
if Cl < 0:
print("ERROR: Remove %i chlorine" % (-Cl))
else:
print("tleap cmd: addionsrand mol Na+ %i Cl- %i 6.0" % (Na, Cl))
To obtain the required number of ions, the script must be executed on the AMBER topology file of the solvated system. For example:
Unload the Amber module and switch to the parmutils environment before
running this command. This avoids syntax warnings that appear when both are
active simultaneously, and because parmutils-GetNumIons.py requires
numpy < 2.
module unload Amber/25.0-foss-2022a-AmberTools-25-CUDA-12.1.1-DeePMD
module purge
source ~/.bashrc
mamba activate parmutils
python3 parmutils-GetNumIons.py MTR1_counterIon_water.parm7
The output of the script includes the recommended tleap command. In this
case, the following line is produced:
tleap cmd: addionsrand mol Na+ 41 Cl- 41 6.0
This indicates that 41 Na+ and 41 Cl- ions must be added to
the system to reach a bulk salt concentration of
0.14 M – on top of the neutralizing part addions mol NA 0 – with a minimum ion–ion separation of 6 Å.
After running parmutils-GetNumIons.py, return to the workshop Amber
environment before continuing:
mamba deactivate
module load Amber/25.0-foss-2022a-AmberTools-25-CUDA-12.1.1-DeePMD
Inclusion of this line completes everything we wanted to do to build our box. Contents of our stand-alone input file are:
set default nocenter on # Do not recenter the molecule
### Sourcing amber forcefields ###############################################
source leaprc.RNA.OL3 # Sourcing RNA force field OL3 to describe RNA.
source leaprc.water.tip4pew # Sourcing water model TIP4P-Ew
# and parameters for corresponding monovalent ions
loadamberparams frcmod.ions234lm_1264_tip4pew # divalent metal ion parameters
### Including non-standard residue parameters ################################
# Ligand O6 methylated guanine
loadoff GUN.lib
loadamberparams GUN.frcmod
# N3 protonated cytosine
loadoff OKC.lib
loadamberparams ./modXNA/dat/frcmod.modxna
### Loading structure from file ##############################################
mol= loadpdb "7v9e_C5.pdb"
### Adding counter ions ######################################################
addions mol NA 0 # Add sodium until charge is zero
### Defining box shape & size with chosen water model ########################
solvateoct mol TIP4PEWBOX 9 # solvate with truncated octahedron box 9A out
### Adding bulk ions to ensure 0.14 M concentration ##########################
addionsrand mol NA 41 CL 41 6.0 # add bulk ions, replacing random waters,
# ensuring 6 A distance between ions
### Saving output files #####################################################
# save parameter and restart files
saveamberparm mol MTR1_counterIon_water_bulk.parm7 MTR1_counterIon_water_bulk.rst7
savepdb mol MTR1_counterIon_water_bulk_amber.pdb
quit
We will use this final input file to build our simulation box.
Run tleap:
tleap -sf MTR1_counterIon_water_bulk.in
This will generate MTR1_counterIon_water_bulk.parm7 and
MTR1_counterIon_water_bulk.rst7 that will be used to perform MD
simulations.
Visually inspect the structure in VMD to get a sense of where ions have been placed.
vmd MTR1_counterIon_water_bulk.parm7 MTR1_counterIon_water_bulk.rst7
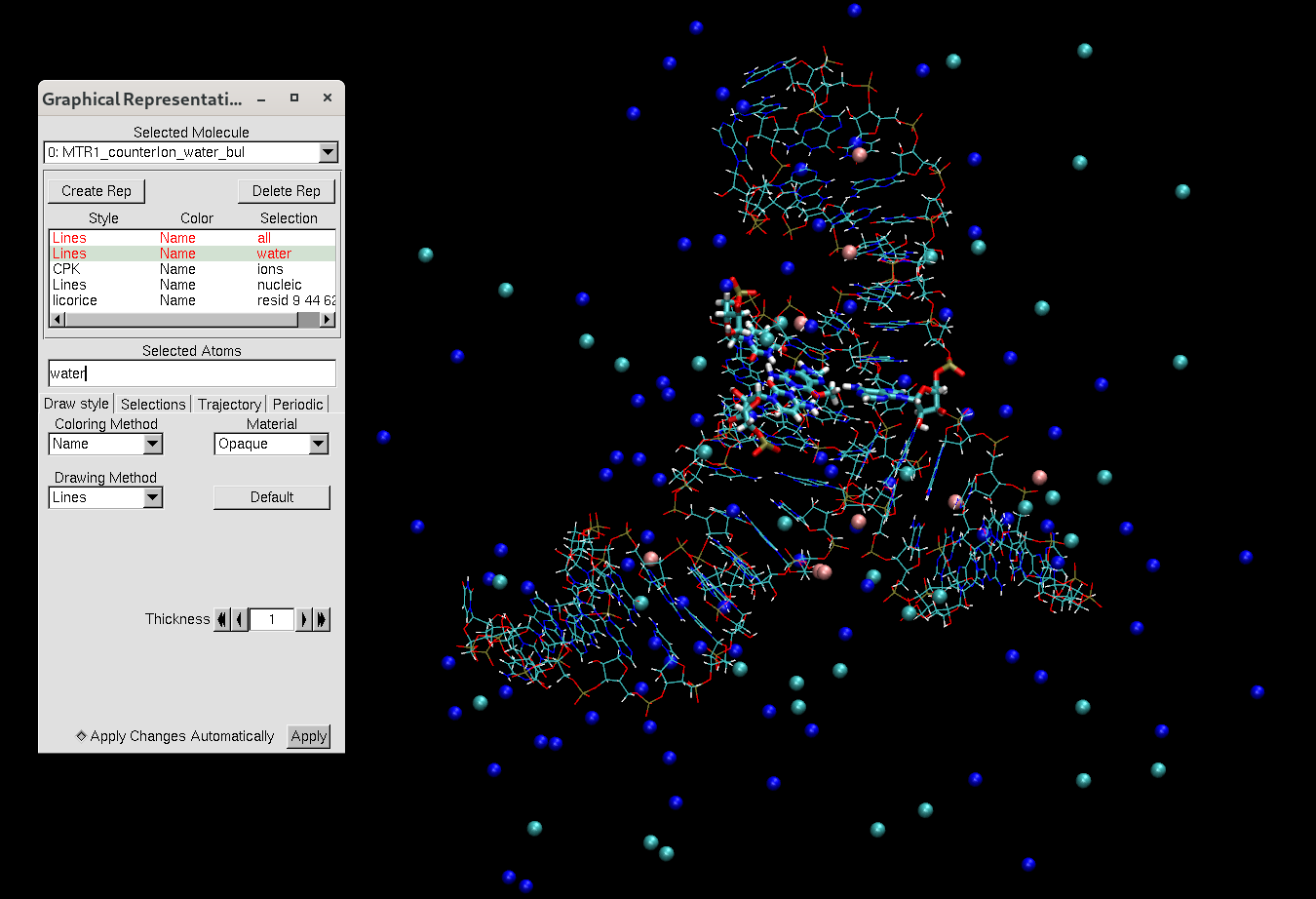
Figure 8. VMD representation of the MTR1 ribozyme with 0.14 M NaCl added to the simulation box.
3.2.5. Divalent metal ion 12-6-4 models with Panteva-Giambasu-York (PGY) pairwise corrections for balanced interactions with nucleic acids
Experimental and theoretical studies have shown that metal ions interact with nucleic acids in two main ways. Electrostatic interactions between the negatively charged nucleic acid backbone and cations lead to a local enrichment of ions around the molecule, known as the ionic atmosphere. In addition to these non-specific interactions, both monovalent and divalent ions can bind more specifically at well-localized sites. In RNA, divalent ions—especially Mg²⁺—can modify local geometry, stabilize tertiary folds, shift the pKa of nearby functional groups, and contribute directly to catalytic activity.
Such binding sites can be identified experimentally using X-ray crystallography, NMR, or spectroscopic techniques. However, these approaches have important limitations: ions are often difficult to identify unambiguously, high and non-physiological ion concentrations are frequently required, and ion positions can be influenced by crystal packing effects. As a result, many questions remain regarding the modes, locations, and strengths of ion binding to nucleic acids, as well as their impact on structure and dynamics. Molecular dynamics simulations therefore provide a valuable complementary approach to study these interactions at the molecular level.
Accurately modeling ion–nucleic acid interactions remains challenging in classical non-polarizable force fields due to the absence of electronic polarization. Fixed atomic charges cannot adapt to the local electrostatic environment, leading to systematic artifacts such as overstabilization of interactions with phosphate groups, underestimation of nucleobase interactions, and an incorrect balance between solvation and direct coordination. As a consequence, divalent ions such as Mg²⁺ may bind too strongly and accumulate unrealistically at the nucleic acid surface.
3.2.5.1. How to overcome these limitations?
One option is to use polarizable force fields, which introduce additional terms into the standard functional form to account for electronic polarization explicitly. Approaches based on induced point dipoles and explicit multipoles provide a more realistic physical description of ion–RNA interactions. However, their higher computational cost and the difficulty of parameterization make them challenging to apply routinely, and they are not yet the preferred choice in large-scale biomolecular simulations.
An alternative is to modify the standard potential to incorporate key polarization effects implicitly. Several models follow this strategy by adjusting Lennard–Jones parameters or adding correction terms. The 12-6-4 model extends the classical Lennard–Jones potential by adding a charge-induced dipole interaction term in \(r^{-4}\):
The coefficients \(A_{ij}\) and \(B_{ij}\) correspond to the usual Lennard–Jones parameters, related to \(R_{ij}\) which is the distance at which two particles have a minimum in the LJ potential, while \(\varepsilon_{ij}\) is the well depth :
The additional coefficient \(C_{ij}\) introduces the charge-induced dipole interaction, which is defined as:
where \(\kappa\) is a scaling parameter (in units of Å⁻²) that determines the strength of the \(r^{-4}\) contribution.
However, although the original 12–6–4 model was developed to accurately reproduce bulk hydration properties, it fails to capture experimentally measured affinities for key RNA-binding sites. This limitation motivated the development of the m12–6–4 model, in which only the \(C_{ij}\) coefficients are re-optimized based on experimental data for three representative sites:
non-bridging oxygens of phosphate
N7 of adenine
N7 of guanine
The pairwise coefficient \(C_{ij}\) for an ion interacting with a given atom type is directly proportional to the polarizability of the atom interacting with the ion \(\alpha\) (in \(\mathrm{Å^3}\)):
This yields several improvements:
more accurate binding free energies between X²⁺-phosphate groups
correct stabilization of nucleobase interactions
an improved balance between solvation and direct coordination
more realistic X²⁺–RNA dissociation kinetics
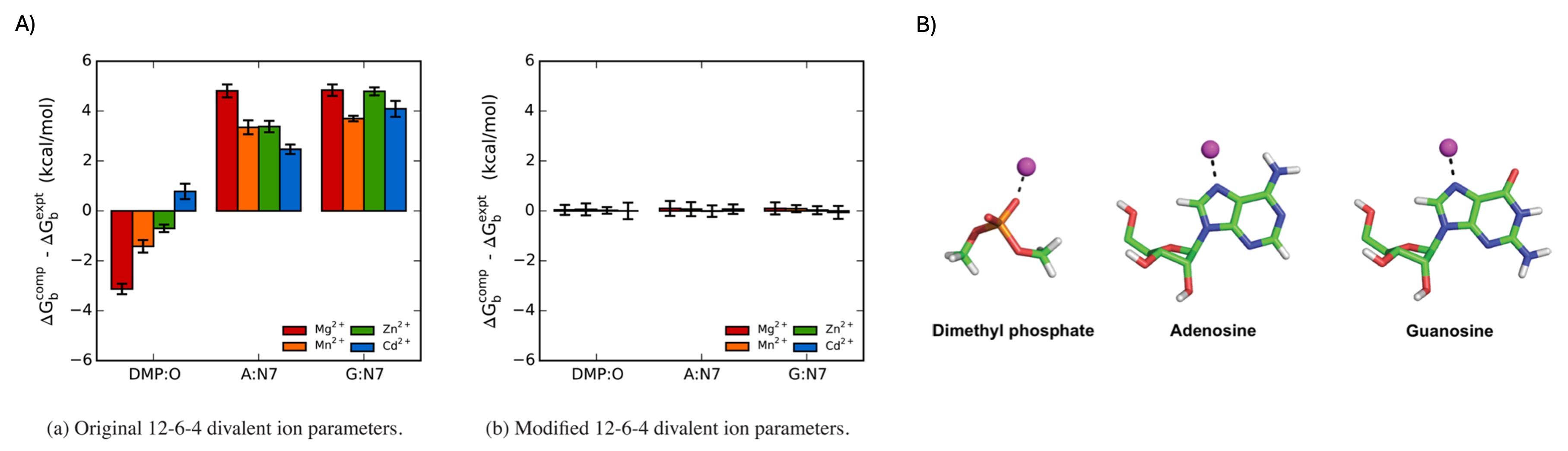
Figure 9. A) Comparison of the errors in the absolute binding free energies for the 12–6–4 (left) and m12–6–4 (right) Mg²⁺, Mn²⁺, Zn²⁺, and Cd²⁺ ion parameters for selected nucleic acid sites. Computed values are averages from three independent simulations, and the error bars correspond to the standard deviations. DMP = dimethyl phosphate, A = adenosine, G = guanosine. B) Model systems and binding sites for which pairwise parameters were tuned to reproduce the reference experimental binding free energies. The magenta sphere represents either Mg²⁺, Mn²⁺, Zn²⁺, or Cd²⁺.
The following files are required to build the 12-6-4 topology and apply the PGY (m12-6-4) pairwise corrections in Amber:
tleap-prep-PDB-2-Amber.in— tleap input file used to generate the initial Amber topology with the 12-6-4 ion model.1264.sh— shell script that prepares the ParmEd instruction file and applies the PGY corrections.parmed_1264_na.in— ParmEd instruction file assigning NAMG/NGMG/OPMG atom types and enabling the PGY corrections.lj_1264_pol.dat— polarizability table used byadd12_6_4to compute the \(C_{ij}\) coefficients.
Here, the goal is to apply these corrections for divalent cations interacting with nucleic acids. Make sure the Amber module is loaded.
We will start from the previously prepared files
MTR1_counterIon_water_bulk.parm7 and
MTR1_counterIon_water_bulk.rst7, which include the 12-6-4 Mg²⁺
model. The m12-6-4 (PGY) corrections are applied in a second pass
using ParmEd.
3.2.5.2. The script 1264.sh
This helper script inserts the proper output file names and runs ParmEd:
#!/bin/bash
# Usage: ./1264.sh SYSTEM
name=$1
cat parmed_1264_na.in > parmed.${name}.in
sed -i '/^outparm/d' parmed.${name}.in
cat <<EOF >> parmed.${name}.in
outparm ${name}_1264.parm7 ${name}_1264.rst7
EOF
parmed -i parmed.${name}.in -p ${name}.parm7 -c ${name}.rst7
Make executable:
chmod +x 1264.sh
3.2.5.3. The file parmed_1264_na.in
This file applies the PGY-specific N7(A), N7(G), and OP corrections:
setOverwrite True
change AMBER_ATOM_TYPE :A*,DA*@N7 NAMG
change AMBER_ATOM_TYPE :G*,DG*@N7 NGMG
change AMBER_ATOM_TYPE :*@OP* OPMG
addLJType @%NAMG
addLJType @%NGMG
addLJType @%OPMG
add12_6_4 @%Mg2+ watermodel TIP4PEW polfile lj_1264_pol.dat
printLJMatrix @%Mg2+
outparm NAME.parm7 NAME.rst7
3.2.5.4. Polarizabilities (lj_1264_pol.dat)
Below is the full content of the lj_1264_pol.dat file:
H 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HC 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H0 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H1 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H2 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H3 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HA 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H4 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
H5 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HO 0.000 Keep its C4 terms consistent with its 12-6 LJ parameters
HS 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HP 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
HZ 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h1 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h2 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h3 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h4 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
h5 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
ha 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
hc 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
hn 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
ho 0.000 Keep its C4 terms consistent with its 12-6 LJ parameters
hp 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
hs 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
hx 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
Hc 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
Ha 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
Ho 0.000 Keep its C4 terms consistent with its 12-6 LJ parameters
Hp 0.387 Referenced or adopted from Miller JACS,112,8533(1990)
C 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C* 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C4 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C5 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CA 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CB 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CC 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CD 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CK 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CM 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CN 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CO 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CP 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CQ 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CR 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CS 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CV 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CW 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CY 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
CZ 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C2 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
C1 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
c 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
c2 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
ca 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cc 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cd 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
ce 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cf 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cp 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cq 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cu 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cv 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cx 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cy 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
cz 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
2C 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
3C 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
C8 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
CI 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
CT 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
CX 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
XC 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
TG 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
c3 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
C7 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
CJ 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
c1 1.283 Referenced or adopted from Miller JACS,112,8533(1990)
cg 1.283 Referenced or adopted from Miller JACS,112,8533(1990)
ch 1.283 Referenced or adopted from Miller JACS,112,8533(1990)
Cg 1.061 Referenced or adoptedfrom Miller JACS,112,8533(1990)
Cy 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
Cp 1.061 Referenced or adopted from Miller JACS,112,8533(1990)
Ck 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
Cj 1.352 Referenced or adopted from Miller JACS,112,8533(1990)
N 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
N* 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
N2 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
N3 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NA 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NB 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NAMG 1.910 From Panteva et al. JPCB, 2015,119,15460
NGMG 1.925 From Panteva et al. JPCB, 2015,119,15460
NAMN 1.770 From Panteva et al. JPCB, 2015,119,15460
NGMN 1.860 From Panteva et al. JPCB, 2015,119,15460
NAZN 1.480 From Panteva et al. JPCB, 2015,119,15460
NGZN 1.640 From Panteva et al. JPCB, 2015,119,15460
NACD 1.580 From Panteva et al. JPCB, 2015,119,15460
NGCD 1.920 From Panteva et al. JPCB, 2015,119,15460
NC 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
ND 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NL 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NT 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
NY 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
TN 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n1 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n2 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n3 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
n4 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
na 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nb 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nc 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nd 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
ne 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nf 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
nh 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
no 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
Ng 1.090 Referenced or adopted from Miller JACS,112,8533(1990)
O 0.569 Referenced or adopted from Miller JACS,112,8533(1990)
O2 0.569 Referenced or adopted from Miller JACS,112,8533(1990)
O3 0.569 Referenced or adopted from Miller JACS,112,8533(1990)
OD 0.569 Referenced or adoptedfrom Miller JACS,112,8533(1990)
OP 0.569 Referenced or adopted from Miller JACS,112,8533(1990)
OPMG 0.170 From Panteva et al. JPCB, 2015,119,15460
OPMN 0.370 From Panteva et al. JPCB, 2015,119,15460
OPZN 0.510 From Panteva et al. JPCB, 2015,119,15460
OPCD 0.680 From Panteva et al. JPCB, 2015,119,15460
OA 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
OH 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
OS 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
OY 0.637 Added by AG
OZ 0.637 Added by AG
OX 0.637 Added by AG
OW 1.444 From "The Structure and Properties of Water" by Eisenberg & Kauzmann
o 0.569 Referenced or adoptedfrom Miller JACS,112,8533(1990)
oh 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
os 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
ow 1.444 From "The Structure and Properties of Water" by Eisenberg & Kauzmann
Oh 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
Os 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
Oy 0.637 Referenced or adoptedfrom Miller JACS,112,8533(1990)
S 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
SH 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
s 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
s2 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
s4 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
s6 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
sh 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
ss 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
sx 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
sy 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
Sm 3.000 Referenced or adoptedfrom Miller JACS,112,8533(1990)
P 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
p2 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
p3 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
p4 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
p5 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pb 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pc 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pd 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pe 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
pf 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
px 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
py 1.538 Referenced or adoptedfrom Miller JACS,112,8533(1990)
PX 1.538 Added by AG
F 0.32 From Applequist et al. JACS,94,2952(1972)
Cl 1.91 From Applequist et al. JACS,94,2952(1972)
Br 2.88 From Applequist et al. JACS,94,2952(1972)
I 4.69 From Applequist et al. JACS,94,2952(1972)
f 0.32 From Applequist et al. JACS,94,2952(1972)
cl 1.91 From Applequist et al. JACS,94,2952(1972)
br 2.88 From Applequist et al. JACS,94,2952(1972)
i 4.69 From Applequist et al. JACS,94,2952(1972)
Li 0.029 From Sangster & Atwood J. Phys. C 11,1541(1978)
Li+ 0.029 From Sangster & Atwood J. Phys. C 11,1541(1978)
IP 0.2495 From Sangster & Atwood J. Phys. C 11,1541(1978)
Na 0.2495 From Sangster & Atwood J. Phys. C 11,1541(1978)
Na+ 0.2495 From Sangster & Atwood J. Phys. C 11,1541(1978)
K 1.0571 From Sangster & Atwood J. Phys. C 11,1541(1978)
K+ 1.0571 From Sangster & Atwood J. Phys. C 11,1541(1978)
Rb 1.5600 From Sangster & Atwood J. Phys. C 11,1541(1978)
Rb+ 1.5600 From Sangster & Atwood J. Phys. C 11,1541(1978)
Cs 2.5880 From Sangster & Atwood J. Phys. C 11,1541(1978)
Cs+ 2.5880 From Sangster & Atwood J. Phys. C 11,1541(1978)
F- 0.9743 From Sangster & Atwood J. Phys. C 11,1541(1978)
Cl- 3.2350 From Sangster & Atwood J. Phys. C 11,1541(1978)
IM 3.2350 From Sangster & Atwood J. Phys. C 11,1541(1978)
Br- 4.5330 From Sangster & Atwood J. Phys. C 11,1541(1978)
I- 6.7629 From Sangster & Atwood J. Phys. C 11,1541(1978)
Be2+ 0.0067 Calculated from B3LYP/6-311++G(2d,2p)
Cu2+ 0.413 Calculated from B3LYP/6-311++G(2d,2p)
CU 0.413 Calculated from B3LYP/6-311++G(2d,2p)
Ni2+ 0.395 Calculated from B3LYP/6-311++G(2d,2p)
Zn 0.344 Calculated from B3LYP/6-311++G(2d,2p)
ZN 0.344 Calculated from B3LYP/6-311++G(2d,2p)
Zn2+ 0.344 Calculated from B3LYP/6-311++G(2d,2p)
Co2+ 0.447 Calculated from B3LYP/6-311++G(2d,2p)
Cr2+ 0.623 Calculated from B3LYP/6-311++G(2d,2p)
Fe 0.518 Calculated from B3LYP/6-311++G(2d,2p)
FE 0.518 Calculated from B3LYP/6-311++G(2d,2p)
Fe2+ 0.518 Calculated from B3LYP/6-311++G(2d,2p)
Mg 0.048 Calculated from B3LYP/6-311++G(2d,2p)
MG 0.048 Calculated from B3LYP/6-311++G(2d,2p)
Mg2+ 0.048 Calculated from B3LYP/6-311++G(2d,2p)
V2+ 0.620 Calculated from B3LYP/6-311++G(2d,2p)
Mn2+ 0.534 Calculated from B3LYP/6-311++G(2d,2p)
Hg2+ 0.707 Calculated from B3LYP/SDD
Cd2+ 0.427 Calculated from B3LYP/SDD
Ca2+ 0.477 Calculated from B3LYP/6-311++G(2d,2p)
C0 0.477 Calculated from B3LYP/6-311++G(2d,2p)
Sn2+ 3.083 Calculated from B3LYP/SDD
Sr2+ 0.813 Calculated from B3LYP/SDD
Ba2+ 1.496 Calculated from B3LYP/SDD
HW 0.000 Water hydrogen / dummy atom
hw 0.000 Water hydrogen / dummy atom
EP 0.000 Water hydrogen / dummy atom
EPW 0.000 Water hydrogen / dummy atom
EP1 0.000 Water hydrogen / dummy atom
EP2 0.000 Water hydrogen / dummy atom
LP 0.000 Lone pair site
Al3+ 0.031 Calculated from B3LYP/6-311++G(2d,2p)
Fe3+ 0.297 Calculated from B3LYP/6-311++G(2d,2p)
Cr3+ 0.352 Calculated from B3LYP/6-311++G(2d,2p)
Y3+ 0.597 Calculated from B3LYP/SDD
La3+ 1.141 Calculated from B3LYP/SDD
Pr3+ 1.101 Calculated from B3LYP/SDD
Nd3+ 1.047 Calculated from B3LYP/SDD
Sm3+ 0.927 Calculated from B3LYP/SDD
Eu3+ 0.885 Calculated from B3LYP/SDD
Tm3+ 0.675 Calculated from B3LYP/SDD
Lu3+ 0.629 Calculated from B3LYP/SDD
Zr4+ 0.436 Calculated from B3LYP/SDD
U4+ 1.044 Calculated from B3LYP/SDD
Th4+ 1.141 Calculated from B3LYP/SDD
M0 0.000 Treat as zero
3.2.5.5. Output files
Running:
./1264.sh MTR1_counterIon_water_bulk
produces:
MTR1_counterIon_water_bulk_1264.parm7MTR1_counterIon_water_bulk_1264.rst7
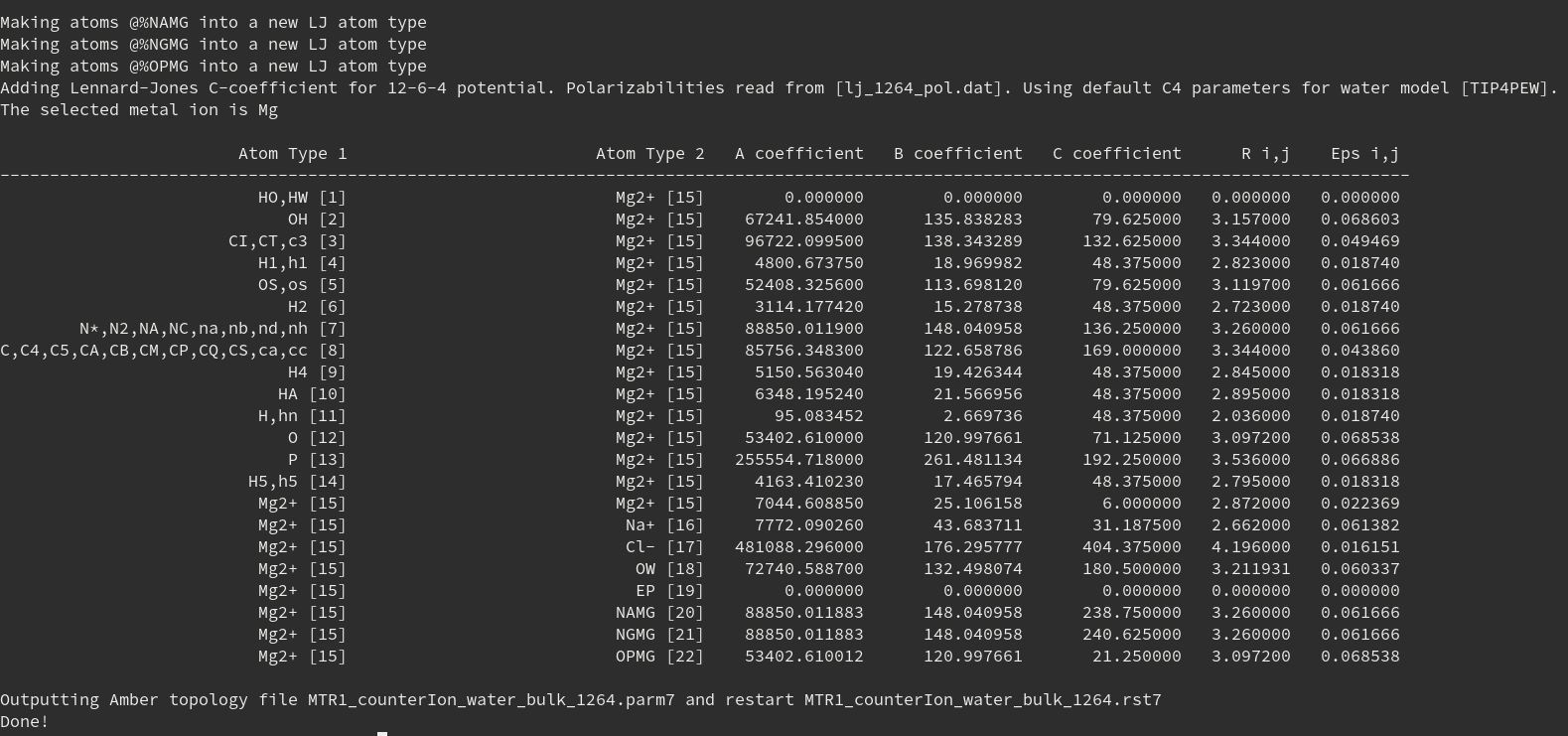
Figure 10. Screenshot of the output showing the interaction matrix involving magnesium.
3.2.6. Optional exercise: derive the GUN ligand parameters
The main workflow above uses the provided GUN.lib and
GUN.frcmod files so that the system can be built without running a
full ligand-parameterization workflow during the hands-on session.
If you would like to see how these files were obtained, the workshop archive also includes the supporting files for this optional exercise:
GUN.pdbGUN.gjfgau.slurmtleap_GUN.inparmed_GUN.in
We next consider the ligand (residue name GUN). Although O6-methylated
guanine is available in modXNA, that implementation assumes the presence
of a nucleic-acid backbone and sugar. In our case, the ligand consists
solely of the base capped with a hydrogen atom; therefore, we use the
standard parametrization approach for non-standard residues.
You will use antechamber to determine atom types, carry out the
restrained electrostatic potential (RESP) procedure to obtain partial
charges for GUN (the O6-methylated guanine ligand), and build
parameter files.
3.2.6.1. Preparing a Gaussian input file
AMBER residue parameters are defined by atom types and partial charges. AMBER has a wide list of atom types with established MM parameters regarding bond lengths, angles, dihedral angles and vdW radii. Partial charges are described separately. Generating parameters for a new residue requires atom type selection and partial charge determination.
In this section you will see how a Gaussian input file was created using GaussView based on the ligand structure in the PDB file. If you have GaussView available, feel free to attempt this section. Otherwise, follow along to see what actions were taken. You have been provided a PDB file of the ligand in the product state. We will need to edit this structure to be in the reactant state.
The following steps were taken to prepare a Gaussian input file for O6 methylated guanine based on the crystal structure coordinates. Recall that the methyl group had already been transferred from the ligand.
If GaussView is available, open GUN.pdb with it:
gv GUN.pdb
A methyl group was added to the oxygen, and the hydrogen placed on C10 in the previous section was deleted.
Figure. Structure of O6-methylated guanine edited with GaussView.
The structure was saved as a Gaussian input file called GUN.gjf,
which is provided in the archive. The input file looks like this:
%chk=GUN.chk
# hf/3-21g
Title Card Required
0 1
N 20.05213100 11.86058800 36.02351400
C 20.78663800 12.65045600 36.89290400
N 20.90570600 12.13218500 38.09712300
C 20.20665200 10.92676800 38.02243900
C 19.93136600 9.86610100 38.92113800
O 20.33478900 9.77794100 40.20264300
N 19.21047500 8.81736400 38.51825700
C 18.75247500 8.79396500 37.24439200
N 17.96142200 7.71264700 36.91957400
N 18.94068400 9.71428900 36.27043500
C 19.66323200 10.73955800 36.72590700
H 19.82722600 12.05265800 35.05295000
H 21.20875900 13.60185700 36.57547400
H 17.88964300 7.52051800 35.92518000
H 18.08578800 6.90435000 37.52121100
C 21.11877500 10.85660800 40.73877700
H 20.56584100 11.80695700 40.70657800
H 21.32467200 10.57091700 41.77831600
H 22.06005100 10.98164000 40.18349700
3.2.6.2. Structure optimization and ESP charges with Gaussian
In this section you will optimize the ligand structure and calculate the electrostatic potential (ESP) charges using Gaussian in order to generate non-standard residue parameters using the restrained electrostatic potential (RESP) procedure. The optimization is done at a high level of DFT, and the ESP is computed at the Hartree–Fock level to be consistent with the methods used to generate the standard AMBER charges.
The input file GUN.gjf has been edited to run an optimization followed
by an ESP calculation and looks as follows:
%chk=GUN.chk
%nprocshared=4
%mem=10GB
# opt pbepbe/6-31g(2d,2p)
GUN_opt
0 1
N 20.05213100 11.86058800 36.02351400
C 20.78663800 12.65045600 36.89290400
N 20.90570600 12.13218500 38.09712300
C 20.20665200 10.92676800 38.02243900
C 19.93136600 9.86610100 38.92113800
O 20.33478900 9.77794100 40.20264300
N 19.21047500 8.81736400 38.51825700
C 18.75247500 8.79396500 37.24439200
N 17.96142200 7.71264700 36.91957400
N 18.94068400 9.71428900 36.27043500
C 19.66323200 10.73955800 36.72590700
H 19.82722600 12.05265800 35.05295000
H 21.20875900 13.60185700 36.57547400
H 17.88964300 7.52051800 35.92518000
H 18.08578800 6.90435000 37.52121100
C 21.11877500 10.85660800 40.73877700
H 20.56584100 11.80695700 40.70657800
H 21.32467200 10.57091700 41.77831600
H 22.06005100 10.98164000 40.18349700
--link1--
%chk=GUN.chk
%nprocshared=4
%mem=10GB
#P HF/6-31G* SCF(Conver=6) NoSymm Test Pop=mk IOp(6/33=2) guess=read geom=allcheck GFInput GFPrint
Note
Make sure there is a blank line at the end of the file.
The first section runs the optimization; the second link uses the
optimized structure to calculate the ESP on a grid of points.
IOp(6/33=2) instructs Gaussian to output this information for RESP
fitting later. A gau.slurm file is provided to submit the job on the
cluster:
sbatch gau.slurm
When the job is finished, verify that the last line of GUN.log reports
“Normal termination of Gaussian”.
3.2.6.3. Atom types and partial charges using antechamber and RESP
Use antechamber to assign atom types and fit RESP charges from the
Gaussian ESP output:
antechamber -i GUN.log -fi gout -o GUN.mol2 -fo mol2 -c resp -at gaff -pf y -rn GUN
This creates GUN.mol2. The flags used:
-c resp— read the ESP from the Gaussian output and perform RESP-at gaff— assign GAFF atom types-pf y— remove intermediate files-rn GUN— set the residue name to GUN
The resulting GUN.mol2 contains the RESP point charges and atom types.
Verify that the charges approximately sum to zero:
sed -n -e '9,27p' GUN.mol2 | awk '{print $9}' > charges.dat
awk '{ sum += $1 } END { print sum }' charges.dat
Expected output: -1e-06
3.2.6.4. Building parameter files
Use tleap to generate a .lib file containing the RESP charges:
cat << EOF > tleap_GUN.in
source leaprc.gaff
GUN = loadmol2 GUN.mol2
SaveOff GUN GUN.lib
saveamberparm GUN GUN.parm7 GUN.rst7
quit
EOF
tleap -sf tleap_GUN.in
Use parmed to write the geometric parameters to a frcmod file:
cat << EOF > parmed_GUN.in
parm GUN.parm7
loadRestrt GUN.rst7
writeFrcmod GUN.frcmod
quit
EOF
parmed -i parmed_GUN.in
You now have all parameter files required to build and run the MTR1 system with the protonated cytosine and the methylated guanine.
3.3. Relevant literature
References cited in this tutorial are listed below.