13. Hands-On Session 13: Apply AmberStudio to perform high-throughput AFE simulations
13.1. Learning Objectives
Describe the core components of AmberStudio and the role each plays in building a simulation workflow.
Distinguish between Artifacts, WorkNodes, Flows, and Pipelines, and explain how they combine to define a calculation.
Identify the input files required to set up an AmberStudio ABFE calculation.
Parameterize a ligand using AmberStudio’s BCC charge workflow.
Build and prepare the topology and coordinate files for an ABFE calculation on Tyk2 inhibitors.
Configure a free (unrestrained) MD calculation using AmberStudio.
Run a system equilibration through an AmberStudio Flow.
Connect multiple Flows together to assemble a multi-stage simulation workflow.
Set up an absolute binding free energy (ABFE) calculation for a set of Tyk2 ligands using the
FlowABFEProductionworkflow.Append an ABFE production flow to the end state of a prior equilibration run.
Run and monitor ABFE calculations for multiple ligands with AmberStudio.
13.2. Activities
flowchart LR
%% ===== generate Binder Parameters =====
A1["binder_ejm31.pdb"]
P1{{"<b/>[§13.2.3]</b><br/>run_bcc_param.py<br/>WorkNode: ParametrizeBinderBCC<br/>amtechamber"}}
O1["<b/>from [§13.2.3]</b><br/>binder_ejm31.mol2<br/>binder_ejm31.lib<br/>binder_ejm31.frcmod<br/>binder_ejm31.pdb"]
A1 --> P1
P1 --> O1
%% ===== Building System =====
A2["target_ejm31.pdb"]
P2{{"<b/>[§13.2.4]</b><br/>run_build.py<br/>Flow: FlowBuild<br/>programs: pdb4amber pdbfixer tleap"}}
O2["<b/>from [§13.2.4]</b><br/>leaf_build output<br/>binder_ejm31.parm7<br/>binder_ejm31.rst7<br/>binderref_ejm31.rst7<br/>complex_ejm31.parm7<br/>complex_ejm31.rst7<br/>complexref_ejm31.rst7"]
O1 --> P2
A2 --> P2
P2 --> O2
%% ===== Equilibrating System =====
P3{{"<b/>[§13.2.5.1]</b><br/>run_equil.py<br/>Flow: FlowRoeRelax<br/>programs: pmemd pmemd.cuda"}}
O3["<b/>[from §13.2.5.1]</b><br/>leaf_roerelax output<br/>binder/complex topology files<br/>equilibrated structures<br/>last-step trajectories (.nc)<br/>mdouts"]
O2 --> P3
P3 --> O3
%% ===== Append FreeMD =====
P4{{"<b>[§13.2.5.2]</b><br/>pmemd.cuda"}}
O4["<b>from [§13.2.5.2]</b><br/>freemd output<br/> binder_ejm31.parm7 binder_ejm31.rst7 binderref_ejm31.rst7 complex_ejm31.parm7 complex_ejm31.rst7 complexref_ejm31.rst7 rest.in"]
O3 --> P4
P4 --> O4
%% ===== ABFE production =====
P5{{"<b>[§13.2.6]</b><br/>run_abfe.py<br/>Flow: FlowABFEProduction<br/>programs: pmemd pmemd.cuda edgembar"}}
O5["<b>from [§13.2.6]</b><br/>ABFE production outputs<br/> leaf abfeproduction - edge_ejm31.html edge_ejm31.nc edge_ejm31.py "]
O4 --> P5
P5 --> O5
classDef input fill:#fff7e6,stroke:#d98c00,stroke-width:1.5px,color:#111;
classDef program fill:#e8f1ff,stroke:#1f77b4,stroke-width:1.8px,color:#111;
classDef output fill:#eaf7ea,stroke:#2ca02c,stroke-width:1.5px,color:#111;
class A1,A2,A3,A4 input;
class P1,P2,P3,P4,P5 program;
class O1,O2,O3,O4,O5 output;
13.2.1. Accessing the Tutorial Files
To get the tutorial data during the workshop, you can get a copy into your scratch with this command.
DataDir="/data/amber26/TutorialData/HandsOn13_AmberStudio"
TutorialDir="/scratch/$USER/TutorialData/HandsOn13_AmberStudio"
mkdir -p $TutorialDir
cp -r $DataDir/* $TutorialDir
cd $TutorialDir
cp -r input working_dir
cd working_dir
The software you will be using for this tutorial is AmberStudio, which is a new (soon to be publicly released) platform for setting up and running amber simulations.
To load it on hyperion, you can use the following command:
source /scratch/amber26/software/flowrc.sh
13.2.2. Introduction to AmberStudio
AmberStudio is a platform for running Molecular Dynamics simulations with Amber [1]. One of the challenges with running complex calculations with Amber is the need to run calculations in many stages (e.g. minimization, density equilibration, heating, equilibration, production) often resulting in more than ten MD input files for a single calculation. AmberStudio allows you to generate custom workflows that automate a significant portion of this process.
We will briefly outline the basic components of AmberStudio.
13.2.2.1. Artifacts
Artifacts are the basic file units passed between WorkNodes. These are usually file-type objects (e.g. ligand topologies, PDB files, NC trajectory files, analysis HTML files).
Below you can see an example of a BinderLigandPDB artifact (which is a PDB of a ligand that binds to a protein).
from amberstudio.artifacts import BinderLigandPDB
artifact = BinderLigandPDB("binder_ejm31.pdb")
print(artifact.filepath)
13.2.2.2. WorkNodes
WorkNodes are the basic unit of work within AmberStudio. Generally, each WorkNode performs one task, transforming its input Artifact objects into output Artifact objects.
In the example below, you can see a WorkNode that applies PDB4Amber to a protein PDB.
This WorkNode takes an input PDB file, applies PDB4Amber to it, and then produces a cleaned PDB as an output Artifact.
from amberstudio.worknodes import PDB4Amber
worknode = PDB4Amber(
wnid="clean_target",
nohyd=True,
skippable=True,
)
print(worknode.id)
WorkNodes specify which types of input artifacts they expect and which type of output artifact they produce. A WorkNode that runs MD expects a topology and a restart file. If, for example, it gets a PDB instead, it will fail.
13.2.2.3. Flows
Flows are pre-defined combinations of WorkNodes that represent protocols. Examples of Flows are System Building, End-State Equilibration, and Absolute Binding Free Energies.
Below, an example for FlowBuild is shown which takes input binder and target PDBs and generates a solvated and neutralized system (.parm7 and .rst7).
from amberstudio.flows import FlowBuild
flow = FlowBuild(
ligand_charge=-1,
skippable=True,
)
print(flow.name)
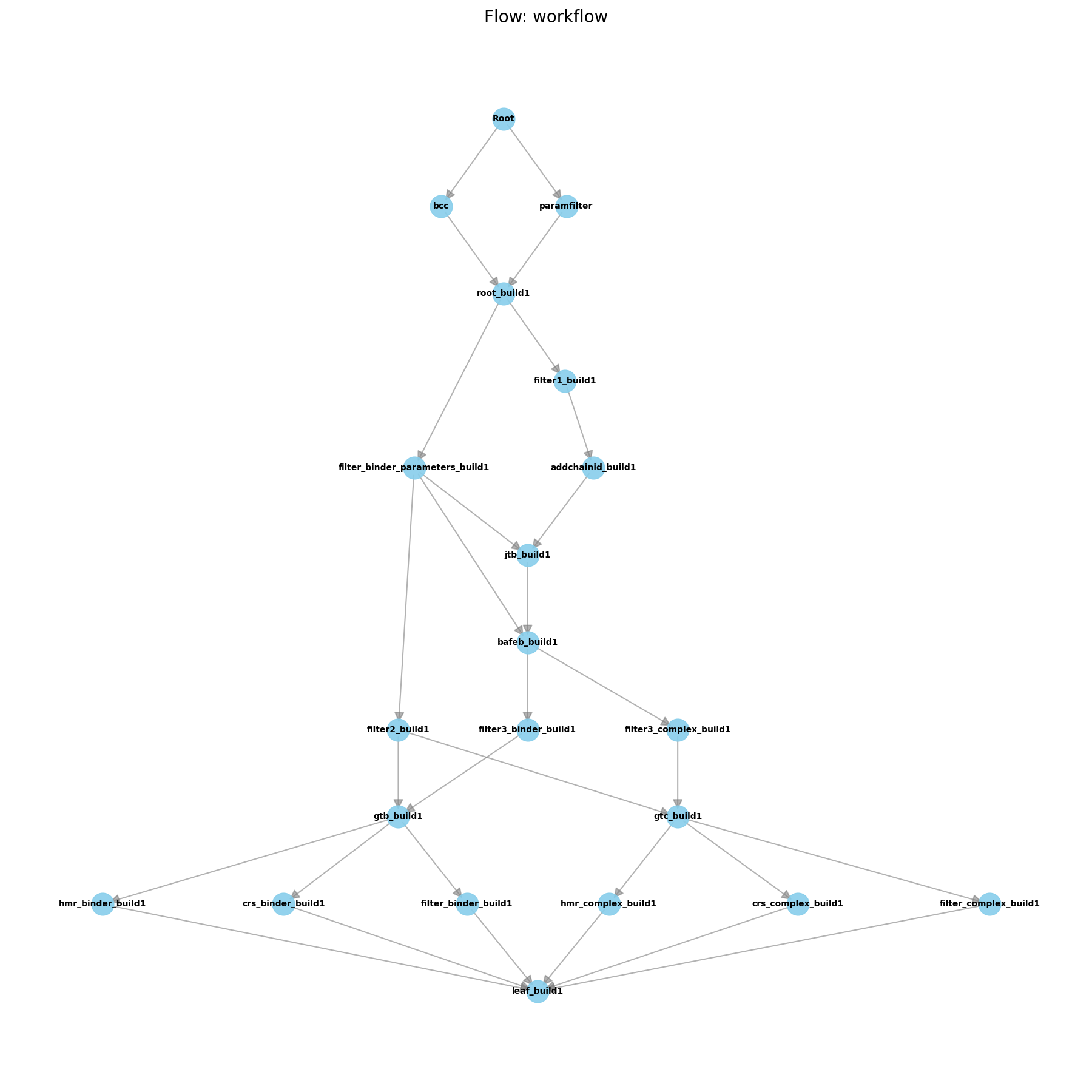
Each node in the graph is a WorkNode, while the edges represent the work dependencies between the WorkNodes and also
take the artifacts from one WorkNode to the next.
13.2.2.4. Schedulers
Schedulers decide how WorkNodes get executed, depending on the available resources. To do this, they need information about the platform they will be running on:
from amberstudio.schedulers import DefaultScheduler
scheduler = DefaultScheduler(max_cores=20, max_gpus=4, allow_gpu_oversubscription=False),
Currently, AmberStudio has three built-in schedulers:
ReferenceScheduler: Executes each WorkNode on each system, one at a time, even if they could be run in parallel. Good for debugging.
DefaultScheduler: Tries to execute each WorkNode on all available systems concurrently, if resources allow.
InterleavedScheduler: Tries to execute all ready-to-run WorkNodes on all systems, if resources allow.
13.2.2.5. Pipelines
The user-facing side of AmberStudio is typically a Pipeline. The pipeline is the context of the whole run. It groups WorkNodes, artifacts, schedulers, and other components in order to orchestrate their execution.
Below is an example for a simple pipeline that calls the build flow.
from amberstudio import Pipeline
from amberstudio.flows import FlowBuild
from amberstudio.schedulers import ReferenceScheduler
pipeline = Pipeline(
"intro_demo",
cwd="systems",
scheduler=ReferenceScheduler(max_cores=1, max_gpus=1, allow_gpu_oversubscription=True),
ignore_checkpoint=True,
)
pipeline.append_flow(FlowBuild())
print(pipeline.name)
To launch the pipeline, you would run the following:
pipeline.launch()
Note
Multiple flows can be appended to the same pipeline if their input/output artifacts match up.
For this tutorial, you will be using the param and build_example subdirectories.
13.2.3. Generating Ligand Parameters with AmberStudio
To generate ligand parameters with AmberStudio, you need to start with a PDB. AmberStudio contains Flows for ligand parameterization that use the Ligand Parameterization tools [2] described in the Ligand Parameterization tutorials located here: Using LigandParam to Parameterize Ligands
To do this with AmberStudio, you would start with the following directory structure:
<top-dir>
run_bcc_param.py
binder_ejm31.pdb
The contents of run_bcc_param.py are as follows:
from pathlib import Path
from amberstudio.artifacts import ArtifactContainer, BinderLigandPDB
from amberstudio.worknodes import ParametrizeBinderBCC
def main() -> None:
cwd = Path(__file__).resolve().parent
ligand_pdb = cwd / "binder_ejm31.pdb"
node = ParametrizeBinderBCC(
"param_bcc",
root_dir=cwd,
resname="LIG",
atom_type="gaff2",
charge_model="bcc",
charge=0,
)
input_artifacts = ArtifactContainer("ejm31", [BinderLigandPDB(ligand_pdb)])
node.run(input_artifacts, cwd=cwd, sysname="ejm31")
if __name__ == "__main__":
main()
If you look at the output WorkNode folder (param_bcc), you will see all the files from the parameterization. The files you would want are:
These would be used to build a system.
Note
In this section, we demonstrated how a single WorkNode could be run independently from a Pipeline;
however, we would rarely want to do this. If we had added this WorkNode to a Pipeline, we wouldn’t
have to specify the ArtifactContainer, and the WorkNode would run over all systems.
13.2.4. Building Systems with AmberStudio
Now, we will demonstrate building a system with AmberStudio.
<top-dir>
run_build.py
<ejm31>/
binder_ejm31.mol2
binder_ejm31.lib
binder_ejm31.frcmod
binder_ejm31.pdb
target_ejm31.pdb
The input files here are:
The ligand parameter files (mol2, lib, frcmod) generated from a ligand parameterization (e.g. using the above script)
binder_ejm31.pdb is a PDB of the ligand.
target_ejm31.pdb is a PDB of the protein target.
Note
AmberStudio assumes that the coordinates of binder_ejm31.pdb correspond to a docked structure. Presently, AmberStudio does not perform docking.
from pathlib import Path
import logging
from amberstudio import Pipeline
from amberstudio.flows import FlowBuild
from amberstudio.schedulers import ReferenceScheduler
def main() -> None:
cwd = Path(__file__).resolve().parent
pipeline = Pipeline(
"ejm31_build",
cwd=cwd,
scheduler=ReferenceScheduler(max_gpus=0),
logging_level=logging.INFO,
ignore_checkpoint=True,
)
pipeline.append_flow(
FlowBuild(
parametrize=False,
skippable=False,
)
)
pipeline.launch()
if __name__ == "__main__":
main()
Note a few things about this script:
We set up a pipeline this time, which contains the build flow.
Since we already parameterized the ligands, we set parametrize to False.
This script brings you from the above files, all the way to a built parm7 and rst7 pair for both the aqueous (ligand in water) and complex configurations (ligand in protein) of the ligand.
sbatch run_build.slurm
Now let’s explore the ejm31 folder. You will see that there are many different folders that have been added. Each folder was the working directory of a separate WorkNode in the calculation.
The leaf_build folder contains the output systems.
Note
These leaf WorkNodes are called “dummy” WorkNodes because they don’t usually do anything but collect the output from a flow into a single directory. Pipelines do the same when they start, they collect all input files into a Root WorkNode.
Try opening these with VMD on your local machine.
vmd python complex_ejm31.parm7 complex_ejm31.rst7
You should see the ligand bound into the protein. In the current example, HMR was applied to the system to use hydrogen-mass repartitioning [3]. No Molecular Dynamics has been run yet.
For this module, you will be using the equil_example subdirectory.
13.2.5. Running Free-MD with AmberStudio
13.2.5.1. System Equilibration
Let’s consider that you have built a protein-ligand complex (e.g. through the system build tutorial), and you want to run free MD on that built system. The first thing needed is to run an equilibration on that system.
We will first prepare a new simulation directory to run this calculation.
<equil-dir>/
run_equil.py
ejm31/
binder_ejm31.parm7
binder_ejm31.rst7
binderref_ejm31.rst7
complex_ejm31.parm7
complex_ejm31.rst7
complexref_ejm31.rst7
You can either download this folder or generate it by copying the files from the leaf_build folder you used previously.
Note
A third option would be to append the equilibration flow in run_equil.py to the previous build pipeline script to run both one after another; however, the present tutorial will assume you are starting from a built topology.
The contents of run_equil.py are as follows.
from pathlib import Path
from amberstudio import Pipeline
from amberstudio.flows import FlowRoeRelax
from amberstudio.schedulers import ReferenceScheduler
def main() -> None:
cwd = Path(__file__).resolve().parent
pipeline = Pipeline(
"ejm31_equil",
cwd=cwd,
scheduler=ReferenceScheduler(
max_cores=20,
max_gpus=1,
allow_gpu_oversubscription=False,
),
logging_level=10,
ignore_checkpoint=True,
)
pipeline.append_flow(
FlowRoeRelax(
complex_full_restraint_mask=":1-178",
complex_noh_restraint_mask=":1-178&!@H=",
binder_full_restraint_mask=":179",
binder_noh_restraint_mask=":179&!@H=",
strong_restraint_wt=50,
bb_restraint_mask="@CA,N,C | :1&!@H=",
bb_restraint_wt=5,
nstlim=120,
nstlim_density_equilibration=120,
skippable=True,
)
)
pipeline.launch()
if __name__ == "__main__":
main()
Warning
This will run an equilibration on a GPU. Make sure you are on an interactive GPU node prior to launching.
Note
For the purposes of this tutorial, we set nstlim=120 to keep the MD calculation short. Much longer runs are needed for actual equilibration.
sbatch run_equil.slurm
Now, if things worked, you should see a short MD run complete. If you cd into the ejm31 folder, you will see many more folders, each with _roerelax (the name of the flow) appended to their names. Each of these corresponds to an MD stage that roughly corresponds to minimization -> heating -> density equilibration -> lower restraints on protein -> short free MD.
The output files are located in leaf_roerelax. You’ll note these are the mdouts, the trajectory from the last step, the topology and an equilibrated structure.
13.2.5.2. Appending Free-MD
Now, we could do the same thing as we did at the start of this tutorial: make a new directory, copy the files from leaf_roerelax, and then start a new pipeline. Instead, let’s just modify the existing pipeline to run free MD.
We have to make a few changes to our python script.
At the top of the file, add
from amberstudio.worknodes import MDParameters, MDRun
Then, between the FlowRoeRelax append and pipeline.launch(), add
filterinputcomplex = Filter(
wnid="filterinputcomplex",
artifact_types=(
BaseComplexTopologyFile,
BaseComplexStructureFile,
BaseComplexStructureReferenceFile,
),
skippable=True,
fail_if_no_artifacts=True,
single=True,
)
pipeline.append_node(filterinputcomplex)
extra_free_md = MDRun(
wnid="final_complex_freemd",
mdin_template="md",
mdparameters=MDParameters(
nstlim=120,
ntpr=10,
dt=0.004,
gamma_ln=5.0,
ntwx=10,
iwrap=0,
nscm=0,
),
)
pipeline.append_node(extra_free_md)
sbatch run_equil_plus_free.slurm
Here we first create a filter for the protein-ligand topology (that acts on leaf_roerelax) and then append an MDRun WorkNode. Again, we use a short run time for simplicity. If you look at the final_complex_freemd folder, you will see that you have a completed MD run.
For this module, you will be using the abfe_example subdirectory.
13.2.6. ABFE with AmberStudio
To run an ABFE calculation with AmberStudio, the default workflow is FlowABFEProduction. This can be appended to the original equilibration flow that we used in the system builds tutorial; however, in this tutorial we will start from the end state of that tutorial.
Download the ABFE example files
Your starting folder will look like this:
<abfe-run-dir>/
run_abfe.py
ejm31/
binder_ejm31.parm7
binder_ejm31.rst7
binderref_ejm31.rst7
complex_ejm31.parm7
complex_ejm31.rst7
complexref_ejm31.rst7
ejm42/
binder_ejm42.parm7
binder_ejm42.rst7
binderref_ejm42.rst7
complex_ejm42.parm7
complex_ejm42.rst7
complexref_ejm42.rst7
jmc27/
binder_jmc27.parm7
binder_jmc27.rst7
binderref_jmc27.rst7
complex_jmc27.parm7
complex_jmc27.rst7
complexref_jmc27.rst7
jmc28/
binder_jmc28.parm7
binder_jmc28.rst7
binderref_jmc28.rst7
complex_jmc28.parm7
complex_jmc28.rst7
complexref_jmc28.rst7
Note that we will actually be running four separate systems here all with the same pipeline.
The full contents of run_abfe.py are included below. Most are reproduced from the earlier tutorial; however, the new addition is FlowABFEProduction.
from pathlib import Path
from amberstudio import Pipeline
from amberstudio.flows import FlowABFEProduction, FlowBuild, FlowRoeRelax
from amberstudio.schedulers import ReferenceScheduler
def main() -> None:
cwd = Path(__file__).resolve().parent
pipeline = Pipeline(
"abfe",
cwd=cwd,
scheduler=ReferenceScheduler(
max_cores=10,
max_gpus=1,
allow_gpu_oversubscription=False,
),
logging_level=10,
ignore_checkpoint=True,
)
flow = FlowABFEProduction(
heating_restraint_mask=":1-290&!@H=",
heating_restraint_wt=5,
complex_restraint_mask=":1-290&!@H=",
complex_restraint_wt=5,
binder_nlambdas=3,
complex_nlambdas=3,
nstlim_free_binder=20,
nstlim_free_complex=20,
nstlim_heat=120,
nstlim_equil1=120,
nstlim_equil2=120,
nstlim_trial=120,
ntpr_trial=120,
ntwx_trial=0,
nmropt=True,
numexchg_binder=5,
numexchg_complex=5,
start_trial_binder=1,
end_trial_binder=1,
start_trial_complex=1,
end_trial_complex=1,
lambda_max_systems=1,
skip_analysis=True,
skippable=True,
)
flow.apply_data_version({"mdin":"amber24"})
pipeline.append_flow(flow)
pipeline.launch()
if __name__ == "__main__":
main()
When you run this, you will see output that tells you that each WorkNode is executing on each of the systems independently.
sbatch run_abfe.slurm
FlowABFEProduction roughly performs the following steps:
Lambda annihilation: Generates lambda windows from the provided lambda schedule.
Equilibration: Equilibrates those lambda windows without replica exchange.
Production: Turns on replica exchange between lambda states.
Analysis: Runs
edgembar-amber2datsandedgembar[4] to produce an HTML report for each ABFE calculation.
As always, the most important output is stored in the leaf node: leaf_abfeproduction. This is where the final analysis is generated, and the edgembar output HTML files are located.