Generating umbrella windows for a 1-dimensional methyl transfer reaction in MTR1
Learning objectives
Generate umbrella windows for a 1D methyl transfer reaction within MTR1
Relevant literature
Introduction
The motivation for using umbrella sampling is that, without restraints, a system undergoing dynamics will very rarely cross a reaction barrier. Even if it does, it will be very sparsely sampled and take much longer than we can afford to sample. Therefore, we apply a series of harmonic biasing potentials, also referred to as an umbrella potential, to force the system over a free energy barrier in the space of the selected reaction coordinate. In this way, we can thoroughly sample the breaking and formation of bonds, or other processes like a conformational change. In order to construct a potential of mean force, we must track the value of the reaction coordinate throughout our simulations so that we can later remove the impact of the bias and compute what the distribution of sampling would look like had we not applied the restraint. This activity will show you how to perform and analyze QM/MM simulations of a methyl transfer reaction in the MTR1 ribozyme using 1 reaction coordinate.
QM region selection
The active site is shown in Figure 1, and includes the methyl donor ligand (O6 methylated guanine), the nucleophillic target A63, and C10 protonated at the N3 position.
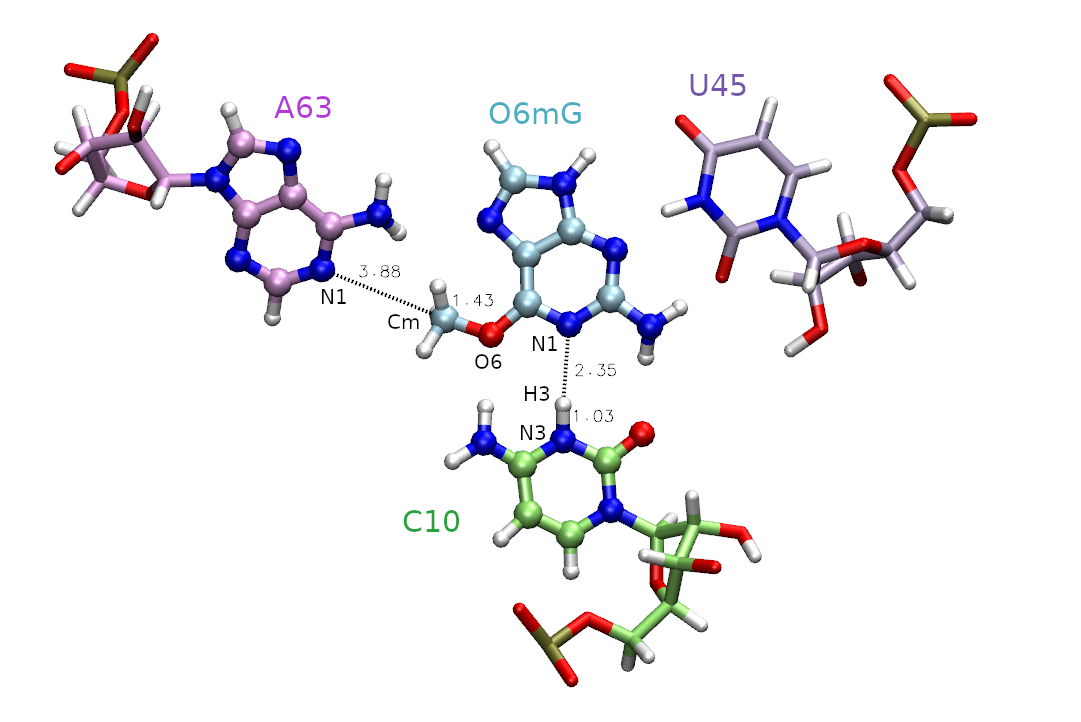
Figure 1. Active site of the MTR1 ribozyme. Atoms included in the QM region are represented as CPK. Distances in are those involved in the methyl transfer reaction coordinate with the associated atoms labeled. The labeled atoms involved in the hydrogen bond between C10 and O6mG will be addressed in part 2.
First, we must decide which atoms will be treated quantum mechanically (aka the QM region). The rest of the atoms will be treated with the molecular mechanical force field. In this case, we are simulating methyl transfer from O6 methylated guanine (O6mG) to the N1 of A63, so we want to include both of these residues in the QM region. The positively charged C10 residue is directly hydrogen bonding to the leaving group, so we will include it as well. In part 2 this residue will also be involved in the reaction, so we will proactively include it for part 1. For A63 and C10, we will only include the nucleobases since the backbones are not directly involved in the reaction. Additionally, U45 is important for holding the ligand in the active site, but since it is not directly involved in the reaction it will remain in the MM region. Based on this selection, the total charge of the QM region is +1 because C10 is in a non-standard protonation state.
Based on this QM region selection, one would need to make some modifications to the parameter file such as removing connection atom charges and redistributing MM charges so that the QM region within each residue is an integer. One would also need to perform hydrogen mass repartitioning in order to run QM/MM simulations stably with a 2 fs time step. These modifications are outside the scope of this exercise and have already been performed to produce qmmm.parm7 that is provided to you in the template directory.
The inputs for this section are located in 1D/inputs. They should be copied to your working directory.
[user@cluster] mkdir 1D
[user@cluster] cd 1D
[user@cluster] cp -r /expanse/projects/qstore/amber_ws/tutorials/QMMM_DFTB3_MTR1/1D/inputs/* ./
Preparing the inputs
You have been provided a parameter file (template/qmmm.parm7), and a structure file (start/reactant.rst7) that was previously equilibrated and represent the reactant state of the ribozyme. Additionally, you have been provided a template mdin file (template/short.mdin). This mdin file is valid for QM/MM simulations in general, and is not specific to umbrella sampling.
Take a look at short.mdin:
title
&cntrl
! IO =======================================
irest = 0 ! 0 = start, 1 = restart
ntx = 1 ! 1 = start, 5 = restart
ntxo = 1 ! read/write rst as formatted file
iwrap = 1 ! wrap crds to unit cell
ioutfm = 1 ! write mdcrd as netcdf
imin = 0
ntmin = 1
ntpr = 20
ntwr = 20
ntwx = 0
ntwf = 0
! DYNAMICS =================================
nstlim = 250
dt = 0.002 ! ps/step
ntb = 1 ! 1=NVT periodic, 2=NPT periodic, 0=no box
! TEMPERATURE ==============================
temp0 = 298 ! target temp
gamma_ln = 5.0 ! Langevin collision freq
ntt = 3 ! thermostat (3=Langevin)
! PRESSURE ================================
ntp = 0 ! 0=no scaling, 1=isotropic, 2=anisotropic
! SHAKE ====================================
ntc = 2 ! 1=no shake, 2=HX constrained, 3=all constrained
noshakemask = ":69|@272-282,290-291,1975-1988" ! do not shake these
ntf = 1 ! 1=cpt all bond E, 2=ignore HX bond E, 3=ignore all bond E
! MISC =====================================
cut = 10.0
ifqnt = 1
ig = -1
nmropt = 1
/
&ewald
dsum_tol = 1.e-6
/
&qmmm
qm_theory = 'DFTB3'
qmmask = ':69|@272-282,290-291,1975-1988'
qmcharge = 1
spin = 1
qmshake = 0
qm_ewald = 1
qmmm_switch = 1
scfconv = 1.e-10
verbosity = 0
tight_p_conv = 1
diag_routine = 0
pseudo_diag = 1
dftb_maxiter = 100
/
&wt type = 'DUMPFREQ', istep1 = 10, /
&wt type='END' /
DISANG=img.disang
DUMPAVE=img.dumpave
LISTOUT=POUT
LISTIN=POUT
As highlighted in cyan, we will be using a 2 fs timestep, but this can only be done safely when hydrogen mass repartitioning has been applied to the parameter file. The initial simulations used to generate the windows for umbrella sampling will only be performed for 0.5 ps.
As highlighted in yellow, we will be using the DFTB3 semi-empirical Hamiltonian, and the total charge of the QM region is +1 due to the protonation of cytosine. When determining the total charge of a nucleic acid QM region, be sure to consider any negatively charged phosphates that may be included. The QM region is defined by the mask ‘:69|@272-282,290-291,1975-1988’ which includes the entire ligand (:69), the nucleobase of C10 (@272-282,290-291) and the nucleobase of A63 (@1975-1988).
The noshakemask (pink) contains the same mask as the QM region, thus we will not shake QM atoms as these bonds involving hydrogen may fluctuate outside of equilibrium MM values when bonds are made/broken. SHAKE constraints are inherently dependent on the MM force field, thus MM constraints should not be combined with our QM description.
Since we want to apply nmr restraints, nmropt (green) is set to 1, and we will output restraint values to the dumpave file every 10 steps. The green highlighted img.disang and img.dumpave are place holders that will be replaced to indicate the appropriate umbrella window number in order to apply restraints.
Let’s review how nmr restraints work in AMBER. When nmropt=1 is set in the mdin file, restraints will be read from the specified DISANG file. These are different than coordinate restraints set when using ntr=1, which effectively freezes the atoms in place. Using nmr restraints allows the atom position to fluctuate subject to a specified biasing potential relating it to another atom or atoms. The values of the restrained properties for a given step are output to the specified DUMPAVE file with a frequency of istep1.
Determine reaction coordinates
Next we need to select the reaction coordinates. We choose the reaction coordinate to be the difference in distance between the atoms where the bond will break, and between the atoms where the bond will form. This ensures the reaction coordinate value to increase going from reactant to product making it more intuitive. In MTR1, describing methyl transfer between O6mG:O6 and A63:N1 will yield the reaction coordinate (RC): RC = |RO6mG:O6 - RCm| - | RA63:N1 - RCm |. We will evaluate the reaction going from the reactant with RC at -2.5 Å, to product with RC at 2.5 Å. You have been provided template/img.disang containing the restraint definitions.
Take a look at img.disang:
#methylation: |GN-C5|-|A62-C5|
&rst iat= 2200, 2189, 2200, 1984,
r1=-999, r2=RC1, r3=RC1, r4=999, rk2=100, rk3=100, rstwt=1.0,-1.0 /
#Max distances for H-bonds
## 1 ##
&rst iat= 1403, 2207,
r1=-999, r2=-999, r3=3.00, r4=999,rk2=0, rk3=100 /
## 2 ##
&rst iat= 1405, 2192,
r1=-999, r2=-999, r3=2.20, r4=999,rk2=0, rk3=100 /
## 3 ##
&rst iat= 1407, 2206,
r1=-999, r2=-999, r3=2.00, r4=999,rk2=0, rk3=100 /
## 4 ##
&rst iat= 2205, 282,
r1=-999, r2=-999, r3=2.20, r4=999,rk2=0, rk3=100 /
## 5 ##
&rst iat= 280, 2189,
r1=-999, r2=-999, r3=2.20, r4=999,rk2=0, rk3=100 /
## 6 ##
&rst iat= 2190, 1982,
r2=-999, r2=-999, r3=3.00, r4=999,rk2=0, rk3=100 /
## 7 ##
&rst iat= 291, 2193,
r1=-999, r2=-999, r3=2.20, r4=999,rk2=0, rk3=100 /
For now, we need to understand more about the restraints used in this example. The first restraint (highlighted yellow) defines the reaction coordinate. The line “&rst iat= 2200, 2189, 2200, 1984,” indicates that we are restraining the difference in distance between the methyl carbon (2200) to ligand oxygen (2189) and the methyl carbon (2200) to the N1 of adenine (1984). These distances are shown in Figure 1. The presence of “rstwt=1.0,-1.0” indicates this should be a difference of distances. In theory, the two distances could be seperate reaction coordinates, but combining them reduces the dimensionality while still describing the same event. The values of r1, r2, r3, and r4 define the shape of the biasing potential. The r2 and r3 values define the flat region of the potential. If r2 is less than r3, then there is no penalty for being within those bounds. When applying an umbrella potential, we set r2=r3 to sample directly on the desired value. The img.disang file is a template, where RC1 will ultimately be replaced for each umbrella window. If more reaction coordinates were present, the placeholder RC2 and so on would be used. The r1 and r4 values define the bounds of the linear response region, ie the penalty will progressively increase as the restrained property approaches r1 and r4. By setting r1=-999 and r4=999 we have effectively created a parabola with infinite bounds. The values rk2 and rk3 are the weights of the restraints. These effectively define the steepness of the parabola, thus controlling the penalty for going outside the r2 to r3 region. For umbrella sampling we want to set this to a high number to force the sampling of high energy regions. The higher the free energy at an umbrella center position, the more the observed value (reported in the dumpave file) will attempt to move away from this center and fight against the biasing potential. It is this concept that helps us construct a PMF.
The rest of the restraints do not define reaction coordinates, and they will be held constant in all of the umbrella windows. These are distance restraints on the active site hydrogen bonds to the ligand to ensure it stays in the pocket. As highlighted in green, these are different from the harmonic potential used for the reaction coordinate because r2=-999 and rk2=0. This defines a half-harmonic biasing potential where any distance less than 3.0 feels no penalty. If the values were reversed (ie. r3=999 and rk2=100), then the two atoms would be pushed away from each other. Now that we have a comprehensive understand of the nmr restraints, we will focus on setting up an umbrella sampling simulation.
Generate the equilibration directory
Now we will generate the equil directory based on the inputs provided. This will be where we sequentially generate the umbrella windows starting from the reactant structure. It is important to slowly increment the reaction coordinate starting from a neighboring structure because each time you change the restraint, you are effectively changing the force field and risk large spikes in energy that could cause unintended bonds to break. We will use the ndfes program to write the inputs for this step.
Navigate to the start directory.
You have been provided a reactant and product.disang file where the reaction coordinate is set to -2.5 and 2.5 respectively. These will define the end points, and a total of 32 umbrella windows will be generated to linearly interpolate through them.
Make sure the amber module is loaded.
Run the following command:
[user@cluster] ndfes-path-prepguess.py --disang ../template/img.disang --mdin ../template/short.mdin --min reactant.disang --min product.disang --nsim 32 --odir ../equil --pad 2 --dry-run
The –min flags indicate the minima we want to use as control points (reactant and product). The option –disang indicates the template disang file with a placeholder, –mdin indicates the template mdin file with a placeholder, –nsim determines the number of umbrella windows, and –pad means the file names will be padded with a leading zero. The –dry-run flag will just print the output without creating the simulation directory called equil. The output should look like this:
1 -2.500 ../equil/img01.disang
2 -2.339 ../equil/img02.disang
3 -2.177 ../equil/img03.disang
4 -2.016 ../equil/img04.disang
5 -1.855 ../equil/img05.disang
6 -1.694 ../equil/img06.disang
7 -1.532 ../equil/img07.disang
8 -1.371 ../equil/img08.disang
9 -1.210 ../equil/img09.disang
10 -1.048 ../equil/img10.disang
11 -0.887 ../equil/img11.disang
12 -0.726 ../equil/img12.disang
13 -0.565 ../equil/img13.disang
14 -0.403 ../equil/img14.disang
15 -0.242 ../equil/img15.disang
16 -0.081 ../equil/img16.disang
17 0.081 ../equil/img17.disang
18 0.242 ../equil/img18.disang
19 0.403 ../equil/img19.disang
20 0.565 ../equil/img20.disang
21 0.726 ../equil/img21.disang
22 0.887 ../equil/img22.disang
23 1.048 ../equil/img23.disang
24 1.210 ../equil/img24.disang
25 1.371 ../equil/img25.disang
26 1.532 ../equil/img26.disang
27 1.694 ../equil/img27.disang
28 1.855 ../equil/img28.disang
29 2.016 ../equil/img29.disang
30 2.177 ../equil/img30.disang
31 2.339 ../equil/img31.disang
32 2.500 ../equil/img32.disang
dmax 0.161
dmax is the spacing between umbrella windows. In general, if the spacing is too larger, consider adding more windows. For our purposes this is sufficiently small.
[user@cluster] ndfes-path-prepguess.py --disang ../template/img.disang --mdin ../template/short.mdin --min reactant.disang --min product.disang --nsim 32 --odir ../equil --pad 2
Generate the umbrella windows
List the contents of the equil directory:
[user@cluster] cd ../equil
[user@cluster] ls
eq01fwd.sh img04.disang img07.mdin img11.disang img14.mdin img18.disang img21.mdin img25.disang img28.mdin img32.disang
img01.disang img04.mdin img08.disang img11.mdin img15.disang img18.mdin img22.disang img25.mdin img29.disang img32.mdin
img01.mdin img05.disang img08.mdin img12.disang img15.mdin img19.disang img22.mdin img26.disang img29.mdin init01.rst7
img02.disang img05.mdin img09.disang img12.mdin img16.disang img19.mdin img23.disang img26.mdin img30.disang
img02.mdin img06.disang img09.mdin img13.disang img16.mdin img20.disang img23.mdin img27.disang img30.mdin
img03.disang img06.mdin img10.disang img13.mdin img17.disang img20.mdin img24.disang img27.mdin img31.disang
img03.mdin img07.disang img10.mdin img14.disang img17.mdin img21.disang img24.mdin img28.disang img31.mdin
You will see that the eq01fwd.sh script was generated for you in the ouputs directory.
Take a look at eq01fwd.sh:
#!/bin/bash
set -e
set -u
#
# You can create a slurm script and run these
# commands in the following way:
#
# export LAUNCH="srun sander.MPI"
# export PARM="path/to/parm7"
# bash eq01fwd.sh
#
if [ "${LAUNCH}" == "" ]; then
echo 'bash variable LAUNCH is undefined. Defaulting to: export LAUNCH="sander"'
export LAUNCH="sander"
fi
if [ "${PARM}" == "" ]; then
echo 'bash variable PARM is undefined. Please: export PARM="/path/to/parm7"'
exit 1
else
if [ ! -e "${PARM}" ]; then
echo "File not found: ${PARM}"
exit 1
fi
fi
if [ ! -e "init01.rst7" ]; then
echo "File not found: init01.rst7"
exit 1
fi
${LAUNCH} -O -p ${PARM} -i img01.mdin -o img01.mdout -c init01.rst7 -r img01.rst7 -x img01.nc -inf img01.mdinfo
${LAUNCH} -O -p ${PARM} -i img02.mdin -o img02.mdout -c img01.rst7 -r img02.rst7 -x img02.nc -inf img02.mdinfo
${LAUNCH} -O -p ${PARM} -i img03.mdin -o img03.mdout -c img02.rst7 -r img03.rst7 -x img03.nc -inf img03.mdinfo
${LAUNCH} -O -p ${PARM} -i img04.mdin -o img04.mdout -c img03.rst7 -r img04.rst7 -x img04.nc -inf img04.mdinfo
${LAUNCH} -O -p ${PARM} -i img05.mdin -o img05.mdout -c img04.rst7 -r img05.rst7 -x img05.nc -inf img05.mdinfo
${LAUNCH} -O -p ${PARM} -i img06.mdin -o img06.mdout -c img05.rst7 -r img06.rst7 -x img06.nc -inf img06.mdinfo
${LAUNCH} -O -p ${PARM} -i img07.mdin -o img07.mdout -c img06.rst7 -r img07.rst7 -x img07.nc -inf img07.mdinfo
${LAUNCH} -O -p ${PARM} -i img08.mdin -o img08.mdout -c img07.rst7 -r img08.rst7 -x img08.nc -inf img08.mdinfo
${LAUNCH} -O -p ${PARM} -i img09.mdin -o img09.mdout -c img08.rst7 -r img09.rst7 -x img09.nc -inf img09.mdinfo
${LAUNCH} -O -p ${PARM} -i img10.mdin -o img10.mdout -c img09.rst7 -r img10.rst7 -x img10.nc -inf img10.mdinfo
${LAUNCH} -O -p ${PARM} -i img11.mdin -o img11.mdout -c img10.rst7 -r img11.rst7 -x img11.nc -inf img11.mdinfo
${LAUNCH} -O -p ${PARM} -i img12.mdin -o img12.mdout -c img11.rst7 -r img12.rst7 -x img12.nc -inf img12.mdinfo
${LAUNCH} -O -p ${PARM} -i img13.mdin -o img13.mdout -c img12.rst7 -r img13.rst7 -x img13.nc -inf img13.mdinfo
${LAUNCH} -O -p ${PARM} -i img14.mdin -o img14.mdout -c img13.rst7 -r img14.rst7 -x img14.nc -inf img14.mdinfo
${LAUNCH} -O -p ${PARM} -i img15.mdin -o img15.mdout -c img14.rst7 -r img15.rst7 -x img15.nc -inf img15.mdinfo
${LAUNCH} -O -p ${PARM} -i img16.mdin -o img16.mdout -c img15.rst7 -r img16.rst7 -x img16.nc -inf img16.mdinfo
${LAUNCH} -O -p ${PARM} -i img17.mdin -o img17.mdout -c img16.rst7 -r img17.rst7 -x img17.nc -inf img17.mdinfo
${LAUNCH} -O -p ${PARM} -i img18.mdin -o img18.mdout -c img17.rst7 -r img18.rst7 -x img18.nc -inf img18.mdinfo
${LAUNCH} -O -p ${PARM} -i img19.mdin -o img19.mdout -c img18.rst7 -r img19.rst7 -x img19.nc -inf img19.mdinfo
${LAUNCH} -O -p ${PARM} -i img20.mdin -o img20.mdout -c img19.rst7 -r img20.rst7 -x img20.nc -inf img20.mdinfo
${LAUNCH} -O -p ${PARM} -i img21.mdin -o img21.mdout -c img20.rst7 -r img21.rst7 -x img21.nc -inf img21.mdinfo
${LAUNCH} -O -p ${PARM} -i img22.mdin -o img22.mdout -c img21.rst7 -r img22.rst7 -x img22.nc -inf img22.mdinfo
${LAUNCH} -O -p ${PARM} -i img23.mdin -o img23.mdout -c img22.rst7 -r img23.rst7 -x img23.nc -inf img23.mdinfo
${LAUNCH} -O -p ${PARM} -i img24.mdin -o img24.mdout -c img23.rst7 -r img24.rst7 -x img24.nc -inf img24.mdinfo
${LAUNCH} -O -p ${PARM} -i img25.mdin -o img25.mdout -c img24.rst7 -r img25.rst7 -x img25.nc -inf img25.mdinfo
${LAUNCH} -O -p ${PARM} -i img26.mdin -o img26.mdout -c img25.rst7 -r img26.rst7 -x img26.nc -inf img26.mdinfo
${LAUNCH} -O -p ${PARM} -i img27.mdin -o img27.mdout -c img26.rst7 -r img27.rst7 -x img27.nc -inf img27.mdinfo
${LAUNCH} -O -p ${PARM} -i img28.mdin -o img28.mdout -c img27.rst7 -r img28.rst7 -x img28.nc -inf img28.mdinfo
${LAUNCH} -O -p ${PARM} -i img29.mdin -o img29.mdout -c img28.rst7 -r img29.rst7 -x img29.nc -inf img29.mdinfo
${LAUNCH} -O -p ${PARM} -i img30.mdin -o img30.mdout -c img29.rst7 -r img30.rst7 -x img30.nc -inf img30.mdinfo
${LAUNCH} -O -p ${PARM} -i img31.mdin -o img31.mdout -c img30.rst7 -r img31.rst7 -x img31.nc -inf img31.mdinfo
${LAUNCH} -O -p ${PARM} -i img32.mdin -o img32.mdout -c img31.rst7 -r img32.rst7 -x img32.nc -inf img32.mdinfo
This script would start from img01 and iteratively generate the umbrella windows using the output of the previous window as the starting structure. It is important to increment the starting structure slowly to avoid errors. One can uses this script to write a slurm script for the desired cluster if necessary.