Mapping Common-Core and Soft-Core Atoms in Relative Binding Free Energy Calculations
Learning objectives
Map common-core and soft-core atoms between two ligands using an atom-mapping tool.
Apply best-practice guidelines for soft-core regions (a single contiguous region, full bond coverage, and limited region size).
Diagnose transformations that violate these guidelines and choose remedies such as intermediate ligands, pseudo-ligands, or CBFE connections.
Introduction to Atom Mapping
Soft-Core Regions for RBFE Calculations
In Relative Binding Free Energy calculations, we are interested in calculating the free energy difference between two ligands binding to the same protein (as opposed to an Absolute Binding Free Energy calculation where we disappear a ligand completely). However, to calculate this free energy difference, we need to alchemically transform one ligand into the other. This transformation is done by identifying a common-core region between the two ligands that is not alchemically transformed, and a soft-core region that is alchemically transformed.
Note
The common-core region is a shared set of atoms present in both ligands that are not alchemically transformed during the simulation.
The soft-core region consists of atoms that are alchemically transformed from one ligand to the other during the simulation. These atoms are typically involved in the chemical differences between the two ligands.
The soft-core region is called “soft-core” because its interactions are lambda dependent and use second-order smoothstep functions to avoid singularities during the alchemical transformation. The choice of which atoms to include in the common-core and soft-core regions can significantly impact the convergence and accuracy of the relative binding free energy calculations.
Mapping Soft-Core and Common-Core Atoms
Before we show any code, let’s first consider some example transformations and how we might choose to map the common-core and soft-core atoms. In this tutorial, we will use an example atom mapping script.
Download the following script to your local machine.
#!/usr/bin/env python3
"""Draw two ligands with RDKit and highlight common-core vs soft-core atoms.
This script is aimed at RBFE tutorials where two ligands are compared as an
alchemical pair. It:
1. loads two ligands from either MOL2 files or SMILES strings
2. finds a maximum common substructure (MCS)
3. identifies each ligand's common-core and soft-core atoms
4. writes ChemDraw-style PNG depictions with those regions highlighted
Example
-------
python rdkit_core_softcore_draw.py \
ejm31/binder_ejm31.mol2 \
ejm42/binder_ejm42.mol2 \
--outdir tutorial_drawings
python rdkit_core_softcore_draw.py \
"CC(=O)Nc1ccc(Oc2ncccn2)cc1" \
"CC(=O)Nc1ccc(Oc2ncc(C)cn2)cc1" \
--name-a ligA \
--name-b ligB
"""
from __future__ import annotations
import argparse
from pathlib import Path
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
from rdkit.Chem import rdFMCS
from rdkit.Chem import rdDepictor
from rdkit.Chem.Draw import rdMolDraw2D
from rdk_amber import element_from_amber_type, load_amber_mol2
COMMON_CORE_COLOR = (0.55, 0.83, 0.55)
SOFT_CORE_COLOR = (0.95, 0.52, 0.52)
def parse_args() -> argparse.Namespace:
parser = argparse.ArgumentParser(
description="Create 2D RDKit drawings with common-core and soft-core highlighting."
)
parser.add_argument(
"ligand_a",
help="First ligand input: MOL2 file path or SMILES string",
)
parser.add_argument(
"ligand_b",
help="Second ligand input: MOL2 file path or SMILES string",
)
parser.add_argument(
"--name-a",
help="Optional display name for ligand A, mainly useful for SMILES input",
)
parser.add_argument(
"--name-b",
help="Optional display name for ligand B, mainly useful for SMILES input",
)
parser.add_argument(
"--outdir",
type=Path,
default=Path("mcss_e2_drawings"),
help="Output directory for SVG and summary files",
)
parser.add_argument(
"--ignore-hydrogens",
action="store_true",
help="Exclude hydrogens from the MCS search and drawings",
)
parser.add_argument("--width", type=int, default=500, help="PNG width in pixels")
parser.add_argument("--height", type=int, default=400, help="PNG height in pixels")
return parser.parse_args()
def mol2_has_amber_atom_types(path: Path) -> bool:
in_atom_section = False
for line in path.read_text(encoding="utf-8").splitlines():
stripped = line.strip()
if stripped.startswith("@<TRIPOS>ATOM"):
in_atom_section = True
continue
if stripped.startswith("@<TRIPOS>") and in_atom_section:
break
if not in_atom_section or not stripped or stripped.startswith("#"):
continue
fields = stripped.split()
if len(fields) < 6:
continue
atom_type = fields[5]
if "." in atom_type:
continue
if element_from_amber_type(atom_type) is not None:
return True
return False
def ligand_name(path: Path) -> str:
stem = path.stem
if stem.startswith("binder_"):
return stem.removeprefix("binder_")
return stem
def sanitize_name(name: str) -> str:
sanitized = "".join(ch if ch.isalnum() or ch in {"-", "_"} else "_" for ch in name)
sanitized = sanitized.strip("_")
return sanitized or "ligand"
def infer_ligand_name(raw_input: str, explicit_name: str | None) -> str:
if explicit_name:
return explicit_name
path = Path(raw_input)
if path.exists():
return ligand_name(path)
return sanitize_name(raw_input[:24])
def load_mol_from_path(path: Path, keep_hydrogens: bool) -> Chem.Mol:
if mol2_has_amber_atom_types(path):
mol = load_amber_mol2(str(path), remove_hs=not keep_hydrogens)
else:
mol = Chem.MolFromMol2File(str(path), removeHs=not keep_hydrogens)
if mol is None:
raise ValueError(f"Failed to read MOL2 file: {path}")
return mol
def load_smiles(smiles: str, keep_hydrogens: bool) -> Chem.Mol:
mol = Chem.MolFromSmiles(smiles)
if mol is None:
raise ValueError(f"Failed to parse SMILES string: {smiles}")
if keep_hydrogens:
mol = Chem.AddHs(mol)
return mol
def load_ligand(raw_input: str, keep_hydrogens: bool, name: str | None) -> tuple[Chem.Mol, str]:
path = Path(raw_input)
ligand_label = infer_ligand_name(raw_input, name)
if path.exists():
mol = load_mol_from_path(path, keep_hydrogens)
else:
mol = load_smiles(raw_input, keep_hydrogens)
mol.SetProp("_Name", ligand_label)
return mol, ligand_label
def find_common_core(mol_a: Chem.Mol, mol_b: Chem.Mol) -> tuple[list[int], list[int], Chem.Mol]:
mcs = rdFMCS.FindMCS(
[mol_a, mol_b],
atomCompare=rdFMCS.AtomCompare.CompareElements,
bondCompare=rdFMCS.BondCompare.CompareOrderExact,
ringMatchesRingOnly=True,
completeRingsOnly=True,
maximizeBonds=True,
timeout=30,
)
if mcs.canceled or not mcs.smartsString:
raise RuntimeError("MCS search failed or timed out without a result.")
query = Chem.MolFromSmarts(mcs.smartsString)
if query is None:
raise RuntimeError("RDKit returned an invalid SMARTS for the common core.")
match_a = list(mol_a.GetSubstructMatch(query))
match_b = list(mol_b.GetSubstructMatch(query))
if not match_a or not match_b:
raise RuntimeError("Could not map the common-core SMARTS back onto both ligands.")
return match_a, match_b, query
def orient_molecules_on_common_core(mol_a: Chem.Mol, mol_b: Chem.Mol, query: Chem.Mol) -> None:
"""Generate matched 2D depictions so the common core keeps one orientation."""
# Use a sanitized ligand as the 2D reference.
# The raw MCS SMARTS query can match substructures, but it may not have ring
# information initialized, which causes depiction matching to fail for
# fused-ring systems in newer RDKit builds.
rdDepictor.Compute2DCoords(mol_a)
rdDepictor.StraightenDepiction(mol_a, minimizeRotation=True)
AllChem.GenerateDepictionMatching2DStructure(
mol_b,
mol_a,
refPatt=query,
acceptFailure=False,
)
def atom_labels(mol: Chem.Mol, atom_indices: list[int]) -> list[str]:
labels = []
for atom_idx in atom_indices:
atom = mol.GetAtomWithIdx(atom_idx)
pdb_name = atom.GetPDBResidueInfo().GetName().strip() if atom.GetPDBResidueInfo() else ""
label = pdb_name or f"{atom.GetSymbol()}{atom_idx + 1}"
labels.append(label)
return labels
def highlight_atom_colors(mol: Chem.Mol, common_core_atoms: list[int]) -> tuple[list[int], dict[int, tuple[float, float, float]]]:
common_core = set(common_core_atoms)
soft_core = [atom.GetIdx() for atom in mol.GetAtoms() if atom.GetIdx() not in common_core]
atom_colors = {idx: COMMON_CORE_COLOR for idx in common_core_atoms}
atom_colors.update({idx: SOFT_CORE_COLOR for idx in soft_core})
return common_core_atoms + soft_core, atom_colors
def draw_combined_png(
mol_a: Chem.Mol,
mol_b: Chem.Mol,
match_a: list[int],
match_b: list[int],
out_path: Path,
width: int,
height: int,
) -> None:
highlight_atoms_a, atom_colors_a = highlight_atom_colors(mol_a, match_a)
highlight_atoms_b, atom_colors_b = highlight_atom_colors(mol_b, match_b)
drawer = rdMolDraw2D.MolDraw2DCairo(width * 2, height, width, height)
opts = drawer.drawOptions()
opts.useBWAtomPalette()
opts.bondLineWidth = 2
opts.fixedBondLength = 32
opts.padding = 0.08
opts.legendFontSize = 18
opts.atomHighlightsAreCircles = False
opts.fillHighlights = True
prepared_mols = [Draw.PrepareMolForDrawing(mol_a), Draw.PrepareMolForDrawing(mol_b)]
drawer.DrawMolecules(
prepared_mols,
legends=[mol_a.GetProp("_Name"), mol_b.GetProp("_Name")],
highlightAtoms=[highlight_atoms_a, highlight_atoms_b],
highlightAtomColors=[atom_colors_a, atom_colors_b],
highlightBonds=[[], []],
)
drawer.FinishDrawing()
out_path.write_bytes(drawer.GetDrawingText())
def write_summary(
path: Path,
mol_a: Chem.Mol,
mol_b: Chem.Mol,
match_a: list[int],
match_b: list[int],
query: Chem.Mol,
) -> None:
core_a = atom_labels(mol_a, match_a)
core_b = atom_labels(mol_b, match_b)
soft_a = atom_labels(
mol_a,
[atom.GetIdx() for atom in mol_a.GetAtoms() if atom.GetIdx() not in set(match_a)],
)
soft_b = atom_labels(
mol_b,
[atom.GetIdx() for atom in mol_b.GetAtoms() if atom.GetIdx() not in set(match_b)],
)
lines = [
f"{mol_a.GetProp('_Name')}: {mol_a.GetNumAtoms()} atoms",
f"{mol_b.GetProp('_Name')}: {mol_b.GetNumAtoms()} atoms",
f"common_core_atoms: {query.GetNumAtoms()}",
f"{mol_a.GetProp('_Name')}_common_core: {', '.join(core_a) if core_a else 'none'}",
f"{mol_b.GetProp('_Name')}_common_core: {', '.join(core_b) if core_b else 'none'}",
f"{mol_a.GetProp('_Name')}_soft_core: {', '.join(soft_a) if soft_a else 'none'}",
f"{mol_b.GetProp('_Name')}_soft_core: {', '.join(soft_b) if soft_b else 'none'}",
]
path.write_text("\n".join(lines) + "\n", encoding="utf-8")
def main() -> None:
args = parse_args()
args.outdir.mkdir(parents=True, exist_ok=True)
keep_hydrogens = not args.ignore_hydrogens
mol_a, name_a = load_ligand(args.ligand_a, keep_hydrogens, args.name_a)
mol_b, name_b = load_ligand(args.ligand_b, keep_hydrogens, args.name_b)
match_a, match_b, query = find_common_core(mol_a, mol_b)
orient_molecules_on_common_core(mol_a, mol_b, query)
png = args.outdir / f"{sanitize_name(name_a)}~{sanitize_name(name_b)}.png"
summary = args.outdir / "common_core_summary.txt"
draw_combined_png(mol_a, mol_b, match_a, match_b, png, args.width, args.height)
write_summary(summary, mol_a, mol_b, match_a, match_b, query)
print(f"Wrote {png}")
print(f"Wrote {summary}")
if __name__ == "__main__":
main()
This script uses the rdk-amber package (a small compatibility library to enable mol2 files with amber atom types to be turned into rdkit molecules [1].)
To use this package, run this (ideally in a conda/other virtual python environment).
pip install rdk-amber
Once your environment is set up, you can download the following mol2 files in a tar.gz format here:
Try running the script on these mol2 files.
tar -xvf atom_mapping.tar.gz
cd mapping
python find_common_core.py binder_ejm31.mol2 binder_jmc27.mol2
This will write a directory with a png image, open that and you should see something like this.
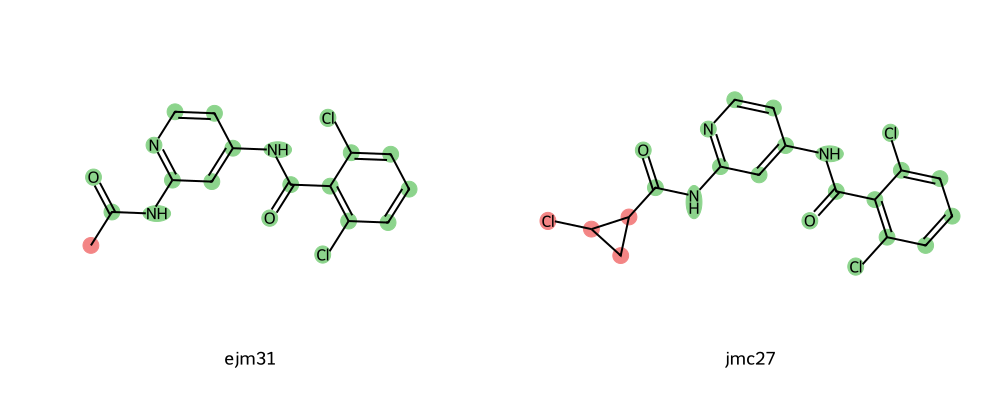
Here, the red highlighted groups are in the soft-core region and the green highlighted groups are in the common-core region.
Try this for the remaining mol2 files - do you notice anything about these soft-core regions?
Considerations for Picking Soft-Core Regions
In general, there are a few best practices to use when generating soft-core regions:
There should only be one soft-core region per transformation (e.g. all red regions with the tool above should be contiguous)
Dihedrals only obtain the benefit of enhanced sampling (dihedral parameters turned off) when soft-core regions fully cover the bond.
If the soft-core region gets too large, then this can negatively impact the replica exchange acceptance ratios.
Now let’s look at a few examples that are not from Tyk2. These come from the paper by Ali Rasouli and co-workers Rasouli et al.[2], which looked at a theophylline-binding RNA aptamer.
We will use the same script as before, but with smiles strings. In the paper, they looked at six molecules that were theophylline-like.
import matplotlib.pyplot as plt
from rdkit import Chem
from rdkit.Chem import Draw
smiles_dict = {
"theophylline": "CN1C2=C(C(=O)N(C1=O)C)NC=N2",
"1-methylxanthine": "CN1C(=O)NC2=C(NC=N2)C1=O",
"3-methylxanthine": "CN1C2=C(NC=N2)C(=O)NC1=O",
"Hypoxanthine": "O=C1NC=Nc2nc[nH]c12",
"Xanthine": "C1=NC2=C(N1)C(=O)NC(=O)N2",
"Caffeine": "CN1C=NC2=C1C(=O)N(C(=O)N2C)C",
}
fig, axes = plt.subplots(2, 3, figsize=(12, 7))
for ax, (name, smiles) in zip(axes.flat, smiles_dict.items()):
mol = Chem.MolFromSmiles(smiles)
img = Draw.MolToImage(mol, size=(300, 220))
ax.imshow(img)
ax.set_title(name, fontsize=12)
ax.axis("off")
fig.tight_layout()
plt.show()
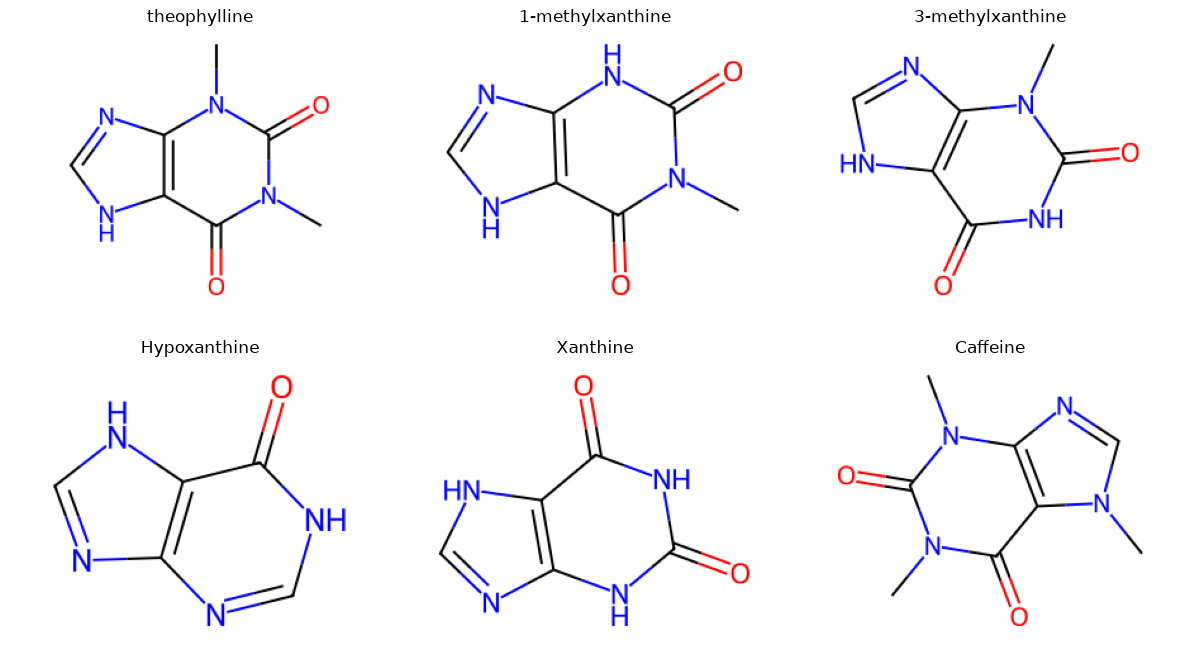
Now let’s call the find_common_core.py script with the SMILES strings for caffeine and xanthine.
python find_common_core.py "C1=NC2=C(N1)C(=O)NC(=O)N2" "CN1C=NC2=C1C(=O)N(C(=O)N2C)C" --name-a xanthine --name-b caffeine
Here, note the name-a and name-b flags make a more convenient output png filename.
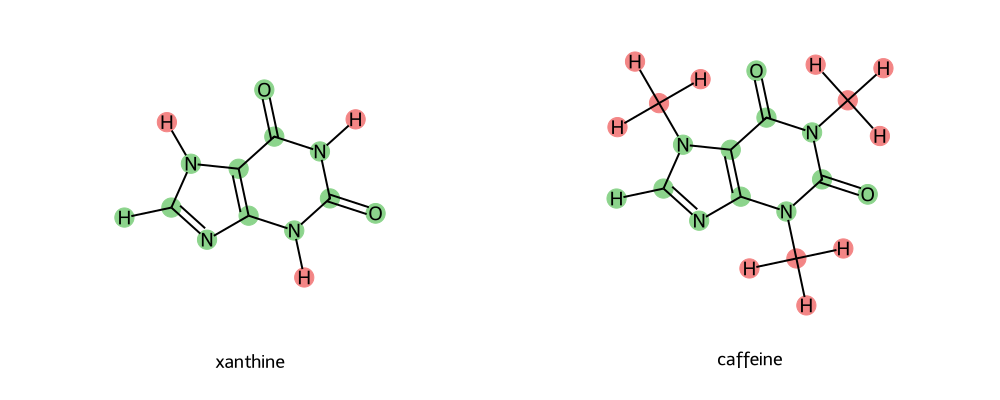
How do these soft-core regions look? Let’s check how they compare to our guidelines above.
Only One Soft-Core Region (FAIL!)
Now - this is a particularly tricky case. You could expand the soft-core region to try to correct this; however, in reality this would essentially engulf the entirety of both molecules. You could try to look for intermediate transformations (however, in this case there is only one valid transformation linking caffeine to a potential network). Can you identify which that is? Try to use the atom-mapping tool to find out. (Answer toggle below)
Caffeine~Theophylline is a valid transformation.
So what would you do?
Three options:
Generate a network, but only connect Caffeine via one edge. Downside: No cycle closure information about caffeine.
Generate a network, but define a pseudo-ligand that bridges the soft-core potential gap. Downside: Can be difficult to generate the molecule, more simulations.
Generate a network, but connect caffeine to other ligands with Counterpoised Binding Free Energy (CBFE) calculations instead. Downside: this is more challenging to resolve and uses a different method.
Note
Counterpoised Binding Free Energies (CBFE)
For cases where it is hard to draw a transformation, or because you want to connect two RBFE networks with differing scaffolds, you can no longer use the RBFE-like infrastructure that we have discussed above. Instead, a more challenging technique is needed: CBFE. Like RBFE, CBFE involves two ligands transforming into one another; however, unlike RBFE, it is a complete transformation without a common core. One ligand is completely disappearing while another ligand is completely appearing. Thus, it is closer to two ABFE calculations run in opposite directions simultaneously. Since both ligands decouple simultaneously, Boresch restraints are often used between the ligands to keep them aligned with one another (as opposed to between the protein and the ligand, as is the case in ABFE).
More information on CBFE can be found in the work by Tsai and co-workers. Tsai et al.[3]